5 Statistical physics
Statistical physics is about macroscopic systems. Classically, the state $r$ of a system of $N$ particles is represented by $2f$ real numbers
\[ \begin{align} r = \left(q_1, \dotsc, q_f;p_1, \dotsc, p_f\right) \end{align} \]
where $f$ is the number of degrees of freedom, $q_i$ are the generalized coordinates and $p_i$ the generalized momenta. Here $\mathcal{O}\left(f\right) = \mathcal{O}\left(N\right)$. Examples of such a system would be a crystal or $10^{23}$ gas particles in a box. These examples make it clear that the microscopic state of a macroscopic system is not relevant. The exact velocity of one of the $10^{23}$ particles does not matter. The microstate is therefore not of interest.
A significant simplification of the statistics is obtained if one assumes a quantum mechanical system. This is because a stationary state in quantum mechanics can be determined by a finite number of discrete quantum numbers if the system is confined in a finite volume. A microstate $r$ can then be defined by a finite number of natural numbers $n_i$,
\[ \begin{align} r = \left(n_i\right). \end{align} \]
A macrostate $M$ is defined by specifying the probabilities $P_r$ for the microstates $r$,
\[ \begin{align} M \coloneqq \left(P_r\right). \end{align} \]
To do this, imagine a number $N$ of identical systems, a so-called ensemble. This can be a mental concept to understand a single system, but it can also be $N$ actually existing systems, such as $N$ spin-containing particles. Let there be $N_r$ systems in the microstate $r$. Then one can define the $P_r$ by
\[ \begin{align} P_r \coloneqq\lim\limits_{N\to\infty}\frac{N_r}{N} \end{align} \]
Now one can assume as limiting conditions that the system is closed, i.e. that the number of particles and energy are constant. In addition, there should be no symmetries from which further restrictive conservation laws would follow. In particular, conservation of momentum and angular momentum should not apply. The density operator of the ensemble is defined by
\[ \begin{align} \newhat{\rho}\left(t\right) \coloneqq\frac{1}{N}\sum_{j = 1}^{N}\left|j\right\rangle\left\langle j\right|. \end{align} \]
Here $j$ runs over all ensemble members and $\left|j\right\rangle$ is the state of the $j-$th system. The states $\left|j\right\rangle$ do not have to be orthogonal to each other, but they must be normalized, in particular several systems can be in the same state. Let $\left(\left|\psi_k\right\rangle\right)$ for $k\geq 1$ be an orthonormal basis of the Hilbert space from which the $\left|j\right\rangle $states come. For the probability $P_l$ of encountering a randomly chosen ensemble member in the state $\left|\psi_l\right\rangle$, one obtains
\[ \begin{align} P_l = \frac{N_l}{N} = \frac{1}{N}\sum_{j = 1}^{N}\left|\left\langle\psi_l|j\right\rangle\right|^2 = \frac{1}{N}\sum_{j = 1}^{N}\left\langle\psi_l|j\right\rangle\left\langle j|\psi_l\right\rangle = \left\langle\psi_l\left|\newhat{\rho}\right|\psi_l\right\rangle. \end{align} \]
Here, $N_r$ was interpreted as the number of systems that are encountered in a quantum mechanical measurement in the state $\left|\psi_r\right\rangle$. The macrostate $\left(P_r\right)$ can be determined from the density operator, which is why the density operator is also called the statistical operator. From this it further follows
\[ \begin{align} \text{tr}\left(\newhat{\rho}\right) &= 1. \end{align} \]
For an expectation value $\left\langle\newhat{A}\right\rangle$, in the case that $\newhat{A}$ is Hermitian, one obtains
\[ \begin{align} \left\langle\newhat{A}\right\rangle &= \frac{1}{N}\sum_{j = 1}^{N}\left\langle j\left|\newhat{A}\right|j\right\rangle = \frac{1}{N}\sum_{j = 1}^{N}\sum_{k = 1}^{\infty}\big\langle j\big|\psi_k\big\rangle\left\langle \psi_k\left|\newhat{A}\right|j\right\rangle = \frac{1}{N}\sum_{j = 1}^{N}\sum_{k = 1}^{\infty}\left\langle \psi_k\left|\newhat{A}\right|j\right\rangle\big\langle j\big|\psi_k\big\rangle\nonumber\\ &= \sum_{k = 1}^{\infty}\Big\langle \psi_k\Big|\frac{1}{N}\sum_{j = 1}^{N}\newhat{A}\Big|j\Big\rangle\Big\langle j\Big|\psi_k\Big\rangle = \sum_{k = 1}^{\infty}\left\langle\psi_k\left|\newhat{A}\newhat{\rho}\right|\psi_k\right\rangle = \text{tr}\left(\newhat{A}\newhat{\rho}\right). \end{align} \]
Let $\left|\psi_l\right\rangle, \left|\psi_m\right\rangle$ be two states; then one has
\[ \begin{align} \left\langle\psi_l|\newhat{\rho}\psi_m\right\rangle &= \frac{1}{N}\langle\psi_l|\sum_{j = 1}^{N}|j\rangle\left\langle j\newvline\psi_m\right\rangle = \frac{1}{N}\sum_{j = 1}^{N}\langle\psi_l|j\rangle\left\langle j\newvline\psi_m\right\rangle = \frac{1}{N}\left(\sum_{j = 1}^{N}\langle\psi_m|j\rangle\left\langle j\newvline\psi_l\right\rangle\right)^\star\nonumber\\ &= \frac{1}{N}\left(\langle\psi_m|\sum_{j = 1}^{N}|j\rangle\left\langle j\newvline\psi_l\right\rangle\right)^\star = \left\langle\psi_m|\newhat{\rho}\psi_l\right\rangle^\star = \left\langle\newhat{\rho}\psi_l|\psi_m\right\rangle. \end{align} \]
The density operator is therefore Hermitian. The last property of the density operator to be noted here is
\[ \begin{align} \text{tr}\left(\newhat{\rho}^2\right) &= \left\langle\newhat{\rho}\right\rangle = \frac{1}{N}\sum_{j = 1}^{N}\left\langle j\left|\newhat{\rho}\right|j\right\rangle = \frac{1}{N}\sum_{j = 1}^{N}\langle j|\frac{1}{N}\sum_{j' = 1}^{N}|j'\rangle\left\langle j'|j\right\rangle\nonumber\\ &= \frac{1}{N^2}\sum_{j, j' = 1}^{N}\left|\left\langle j|j'\right\rangle\right|^2\leq 1\frac{1}{N^2}\sum_{j, j' = 1}^{N}1 = 1. \end{align} \]
Thus $\text{tr}\left(\newhat{\rho}^2\right) = 1$ holds if and only if all ensemble members are in the same state. This is called a pure state, otherwise it is called a mixed state.
The expectation value of the Hamiltonian is the expectation value of the energy, or the energy $E$ for short. Thus one has
\[ \begin{align} E = \text{tr}\left(\newhat{H}\newhat{\rho}\right). \end{align} \]
With the time-dependent SE, it follows
\[ \begin{align} i\hbar\frac{\partial\newhat{\rho}}{\partial t} &= \frac{1}{N}\sum_{i = 1}^{N}\newhat{H}\left|j\right\rangle\left\langle j\right| + \left|j\right\rangle i\hbar\frac{\partial}{\partial t}\left\langle j\right| \end{align} \]
With
\[ \begin{align} i\hbar\frac{\partial}{\partial t}\left\langle j\right| &= i\hbar\frac{\partial}{\partial t}\left| j\right\rangle^+= -\left(i\hbar\frac{\partial}{\partial t}\left| j\right\rangle\right)^+= -\left(\newhat{H}\left| j\right\rangle\right)^+= -\left\langle j\right|\newhat{H} \end{align} \]
one obtains
\[ \begin{align} i\hbar\frac{\partial\newhat{\rho}}{\partial t} = \left[\newhat{H}, \newhat{\rho}\right].\tag{5.14}\label{eq:von_neumann} \end{align} \]
This is the von Neumann equation.
Let a closed system be exposed to a constant perturbation operator $\newhat{v}_h$; then, by Eq. (4.392), for the transition rate $R_{r, r'}$ between two microstates $\left|\psi_r\right\rangle, \left|\psi_{r'}\right\rangle$, one obtains
\[ \begin{align} R_{r, r'} = \frac{2\pi}{\hbar}\left|\left\langle\psi_{r}\left|\newhat{v}_h\right|\psi_{r'}\right\rangle\right|^2 = R_{r', r}. \end{align} \]
For the time derivative of the probabilities $P_r\left(t\right)$, it thus follows
\[ \begin{align} \frac{dP_r\left(t\right)}{dt} = -\sum_{r'}^{}R_{r, r'}P_r + \sum_{r'}^{}R_{r', r}P_{r'} \end{align} \]
Eq. (5.17) is called the master equation. Define
\[ \begin{align} H\left(t\right) \coloneqq\sum_{r = 1}^{\infty}P_r\ln\left(P_r\right). \end{align} \]
Then one has
\[ \begin{align} \frac{dH}{dt} &= \sum_{r}\left(\newdot{P}_r\ln\left(P_r\right) + \newdot{P}_r\right) = \frac{1}{2}\left[\sum_{r}^{}\newdot{P}_r\ln\left(eP_r\right) + \sum_{r'}\newdot{P}_{r'}\ln\left(eP_{r'}\right)\right]\nonumber\\ &= \frac{1}{2}\sum_{r, r'}^{}R_{r, r'}\left(P_{r'} - P_r\right)\left[\ln\left(eP_r\right) - \ln\left(eP_{r'}\right)\right] = \frac{1}{2}\sum_{r, r'}^{}R_{r, r'}P_r\left(1 - \frac{P_{r'}}{P_r}\right)\ln\left(\frac{P_{r'}}{P_r}\right)\tag{5.19}\label{eq:deriv_h_theorem} \end{align} \]
One has $R_{r, r'}P_r\geq 0$, as well as
\[ \begin{align} \left(1 - x\right)\ln\left(x\right)\leq 0. \end{align} \]
Thus one has
\[ \begin{align} \frac{dH}{dt}\leq 0.\tag{5.21}\label{eq:h_theorem} \end{align} \]
Eq. (5.21) is called the H-theorem. The equilibrium state is defined as the state with constant $P_r$, i.e. with $\newdot{H} = 0$ or $H$ minimal. One further defines the entropy $S$ by
\[ \begin{align} S \coloneqq - k_BH \end{align} \]
What can be said about entropy is:
Second Law of Thermodynamics
The entropy $S$ is maximal in equilibrium. For non-equilibrium processes, $\frac{dS}{dt}>0$, whereby a direction of time is singled out. Such a process is therefore irreversible.
5.1 Microcanonical ensemble
According to Eq. (5.19), in equilibrium one has
\[ \begin{align} P_{r} = P_{r'} \end{align} \]
for all $r, r'$ with $E_r = E_{r'}$. In a closed system, by definition, only states with $E_r = E$ can be achieved.
A closed system in equilibrium is equally likely in each of its accessible microstates.
One can now measure the energy $E$ of the system, with an accuracy $\delta E\gg \Delta E_r$, where $\Delta E_r$ represents a typical spacing between the energy levels of the system. The fact that an energy $E$ was measured with the precision $\delta E$ should be interpreted as meaning that the true energy $E'$ of the system lies in the interval $\left[E - \delta E, E\right)$. There are therefore a number of states $r$ whose energies $E_r$ lie in this interval and are therefore compatible with the measurement. One defines the microcanonical partition function $\Omega$ by
\[ \begin{align} \Omega\left(E, x\right) \coloneqq \sum_{r:E - \delta E\leq E_r< E}^{}1. \end{align} \]
Here $x$ denotes all parameters other than the energy that define the system macroscopically, in particular the volume. Since all accessible microstates are equally probable, one has
\[ \begin{align} P_r = \frac{1}{\Omega\left(E, x\right)}\cdot\begin{cases} 1, \:E - \delta E\leq E_r < E,\\ 0, \text{ otherwise} \end{cases}\tag{5.25}\label{eq:mikrokan_ens} \end{align} \]
In the microcanonical ensemble, to which the microcanonical partition function refers, all systems are closed and have the same energies and particle numbers. So these are very specific physical requirements. In this case, for the entropy one obtains
\[ \begin{align} S\left(E, x\right) = -k_B\sum_{r = 1}^{\infty}\frac{1}{\Omega\left(E, x\right)}\ln\left(\frac{1}{\Omega\left(E, x\right)}\right) = k_B\ln\left[\Omega\left(E, x\right)\right].\tag{5.26}\label{eq:entropie_mikrokanonisch} \end{align} \]
For the energy of a system, one can write, with reference to an orthonormal basis $\left(\left|\psi_k\right\rangle\right)$ of eigenstates of the Hamiltonian,
\[ \begin{align} E = \sum_{k = 1}^{\infty}P_kE_k\left(x\right). \end{align} \]
For the change in energy, to linear order, it follows
Here, a partial derivative was simply noted for the derivative with respect to $x$, in general it is a sum of several partial derivatives, and $E_k = E$ was also used. Heat means a change in the occupation probabilities with constant external parameters $x$, Work means a change in the energy due to a change in the external parameters; these are conceptual definitions. A differential $df$ is a shorthand notation for a linear Taylor expansion of $f$ ignoring the error term. Since the energy $E$ as a state variable does not depend on the process that led to the state, without loss of generality a quasi-static processThe word quasi can be translated as almost. can be assumed:
\[ \begin{align} dE = dQ_{\text{qs}} - dW_{\text{qs}}\tag{5.29}\label{eq:td_1_0} \end{align} \]
In such a process a sequence of equilibrium states is passed through; one can imagine it as an infinitely slow process. Furthermore, for each external parameter $x_i$ one defines a generalized force $X_i$ by
\[ \begin{align} X_i \coloneqq - \sum_{k = 1}^{\infty}P_k\frac{\partial E_k\left(x\right)}{\partial x_i}.\tag{5.30}\label{eq:def_gen_kraft} \end{align} \]
In equilibrium this becomes
\[ \begin{align} X_i = -\frac{\partial E\left(x\right)}{\partial x_i}. \end{align} \]
In the case of a quasi-static process, the following applies:
\[ \begin{align} dW_{\text{qs}} = -\sum_{i}^{}\frac{\partial E\left(x\right)}{\partial x_i}dx_i = \sum_{i}^{}X_idx_i.\tag{5.32}\label{eq:work_qs} \end{align} \]
The pressure $p$ is the generalized force of the volume:
\[ \begin{align} p\left(E, x\right) \coloneqq - \sum_{k = 1}^{\infty}P_k\frac{\partial E_k\left(x\right)}{\partial V} \end{align} \]
In the case $x = V$, when the only external parameter is the volume, it follows
The temperature $T$ is defined by
\[ \begin{align} \frac{1}{T} \coloneqq \frac{\partial S\left(E, x\right)}{\partial E}\tag{5.35}\label{eq:def_temperatur} \end{align} \]
furthermore, one sets
\[ \begin{align} \beta \coloneqq\frac{1}{k_BT} \end{align} \]
as an abbreviation. Eq. (5.30) is unwieldy because it has to be averaged over an infinite number of quantum mechanical states. To derive an alternative formula, one starts from the microcanonical partition function
\[ \begin{align} \Omega\left(E, x\right) = \Omega\left(E, x_1, x_i\right) = \sum_{r:E - \delta E\leq E_r<E}^{}1 \end{align} \]
here, $x_i$ stands for all external parameters except $x_1$, where $x_1$ is chosen arbitrarily. One has
\[ \begin{align} \frac{\partial\ln\left[\Omega\left(E, x\right)\right]}{\partial x_1} &= \frac{\ln\left[\Omega\left(E, x_1 + dx_1, x_i\right)\right] - \ln\left[\Omega\left(E, x_1, x_i\right)\right]}{dx_1}. \end{align} \]
Now one computes
\[ \begin{align} \Omega\left(E, x_1 + dx_1, x_i\right) &= \sum_{r:E - \delta E\leq E_r\left(x_1 + dx_1, x_i\right)<E}^{}1 = \sum_{r:E - \delta E\leq E_r\left(x\right) + dE_r<E}^{}1 \end{align} \]
with
\[ \begin{align} dE_r = \frac{\partial E_r}{\partial x_1}dx_1. \end{align} \]
One can set
\[ \begin{align} dE_r = \newoverline{dE_r} \end{align} \]
where the averaging is performed over all microstates of the equilibrium state:
\[ \begin{align} \Omega\left(E, x_1 + dx_1, x_i\right) &= = \sum_{r:E - \newoverline{dE_r} - \delta E\leq E_r\left(x\right)<E - \newoverline{dE_r}}^{}1 = \Omega\left(E - \newoverline{dE_r}, x\right) \end{align} \]
Because of
\[ \begin{align} X_i = -\newoverline{\frac{\partial E}{\partial x_i}} \end{align} \]
one has
\[ \begin{align} \newoverline{dE_r} = -X_1x_1. \end{align} \]
Thus one has
\[ \begin{align} \frac{\partial\ln\left[\Omega\left(E, x\right)\right]}{\partial x_1} &= -\frac{\ln\left[\Omega\left(E - \newoverline{dE_r}, x\right)\right] - \ln\left[\Omega\left(E, x\right)\right]}{\newoverline{dE_r}/X_1} = \frac{\partial\ln\left[\Omega\left(E, x\right)\right]}{\partial E}X_1 = \beta X_1\nonumber\\ \Rightarrow X_i &= T\frac{\partial S\left(E, x\right)}{\partial x_i}. \end{align} \]
For the pressure, it follows
\[ \begin{align} p = T\frac{\partial S\left(E, x\right)}{\partial V}\tag{5.46}\label{eq:pressure_prop_0} \end{align} \]
For the change in entropy, one has
\[ \begin{align} dS &= \frac{\partial S\left(E, x\right)}{\partial E}dE + \sum_{i}\frac{\partial S\left(E, x\right)}{\partial x_i}dx_i = \frac{dE}{T} + \sum_{i}^{}\frac{X_i}{T}dx_i\tag{5.47}\label{eq:diff_entropie}\\ \Leftrightarrow dS &= \frac{1}{T}\left(dQ - dW + dW_{\text{qs}}\right) = \frac{dQ_{\text{qs}}}{T}\tag{5.48}\label{eq:entropie_heat} \end{align} \]
If two systems $A, B$ with the microcanonical partition functions $\Omega_A\left(E_A, x_A\right)$ and $\Omega_E\left(E_B, x_B\right)$ are in equilibrium, then for the energy $E$ of the entire system one has
\[ \begin{align} E = E_A + E_B. \end{align} \]
For the partition function $\Omega\left(E, x\right)$ of the entire system, it follows
\[ \begin{align} \Omega\left(E, x\right) = \Omega_A\left(E_A, x_A\right)\Omega_B\left(E_B, x_B\right). \end{align} \]
The partition functions of the subsystems must be multiplied, since all combinations of microstates can occur. For the entropy $S$ of the entire system, it follows
\[ \begin{align} S\left(E, x\right) = k_B\ln\left[\Omega_A\left(E_A, x_A\right)\right] + k_B\ln\left[\Omega_B\left(E_B, x_B\right)\right] = S_A\left(E_A, x_A\right) + S_B\left(E - E_A, x_B\right). \end{align} \]
So the entropy is additive. In equilibrium it is maximal:
\[ \begin{align} \frac{\partial S}{\partial E_A} = 0\Rightarrow\frac{\partial S_A}{\partial E_A} + \frac{\partial S_B}{\partial E_A} = \frac{\partial S_A}{\partial E_A} - \frac{\partial S_B}{\partial E_B} = 0\Rightarrow T_A = T_B\tag{5.52}\label{eq:t_gleichgewicht} \end{align} \]
If the systems $A, B$ are also in $x_i-$exchange, one obtains the condition under the assumption $x_{i} = x_{i, A} + x_{i, B}$
\[ \begin{align} \nabla S = 0\Rightarrow \frac{\partial S}{\partial x_{i, A}} = 0\Rightarrow \frac{\partial S_A}{\partial x_{i, A}} + \frac{\partial S_B}{\partial x_{i, A}} = \frac{\partial S_A}{\partial x_{i, A}} - \frac{\partial S_B}{\partial x_{i, B}} = 0. \end{align} \]
From this, under the assumption $T_A = T_B$, the equality of the generalized forces follows
An equation of the form
\[ \begin{align} p = p\left(T, V, N\right) \end{align} \]
is called thermal equation of state, while equations of the form
\[ \begin{align} E = E\left(T, V, N\right) \end{align} \]
can be referred to as caloric equations of state. At this point, one points out the notation customary in thermodynamics
\[ \begin{align} \frac{\partial f\left(x, y, z\right)}{\partial x} = \left(\frac{\partial f}{\partial x}\right)_{y, z} \end{align} \]
for partial derivatives. The heat capacity of a substance is defined by
\[ \begin{align} C^{(p)} \coloneqq \left(\frac{dQ_{\text{qs}}}{dT}\right)_{p} \end{align} \]
or
\[ \begin{align} C^{(V)} \coloneqq \left(\frac{dQ_{\text{qs}}}{dT}\right)_{V}, \end{align} \]
depending on whether the pressure or the volume is kept constant during the process. With Eq. (5.48), it follows
\[ \begin{align} C^{(p)} &= T\left(\frac{\partial S}{\partial T}\right)_p, \tag{5.60}\label{eq:waermekap_p_entropie}\\ C^{(V)} &= T\left(\frac{\partial S}{\partial T}\right)_V\tag{5.61}\label{eq:waermekap_v_entropie}. \end{align} \]
The specific heat capacity of a system with mass $m$ is defined by
\[ \begin{align} c^{(p)} \coloneqq \frac{1}{m}C^{(p)} \end{align} \]
and analogously for $c^{(V)}$.
5.2 Canonical and grand canonical ensemble
The canonical ensemble consists of systems $A$ that are in heat exchange with large surrounding systems $B$, so-called bath systems. However, there is no particle or $x_i$ exchange. The entire system consisting of $A$ and $B$ is assumed to be closed, so that the microcanonical ensemble can be used. Then, for the total energy, one has
\[ \begin{align} E = E_A + E_B = \text{const}. \end{align} \]
For the microcanonical partition function $\Omega$ of the entire system, one has
\[ \begin{align} \Omega = \Omega\left(E, x\right). \end{align} \]
The entire system is equally likely to be found in each of its accessible microstates. The number of states $n\left(E_A\right)$ with a fixed energy $E_A$ of the system $A$ is given by
\[ \begin{align} n\left(E_A\right) = \Omega_A\left(E_A, x_A\right)\Omega_B\left(E_B, x_B\right), \end{align} \]
where $\Omega_A$ is the microcanonical partition function of system $A$ and $\Omega_B$ is that of system $B$. The probability of encountering a state $r$ with an energy $E_r = E_A$ in the system $A$ is the probability
\[ \begin{align} P\left(E_A\right) = \frac{n_A}{\Omega\left(E, x\right)} = \frac{\Omega_A\left(E_A, x_A\right)\Omega_B\left(E_B, x_B\right)}{\Omega\left(E, x\right)}, \end{align} \]
to find the system $A$ at an energy $E_A$, divided by the number of these equally probable states:
\[ \begin{align} P_r = \frac{P\left(E_A\right)}{\Omega_A\left(E_A, x_A\right)} = \frac{\Omega_B\left(E - E_A, x - x_A\right)}{\Omega\left(E, x\right)} \end{align} \]
$E_B = E - E_A$ and $x_B = x - x_A$ were inserted. Because of $E_B\gg E_A$, one can expand the numerator in $E_A$ about $E_A = 0$:
\[ \begin{align} \ln\left[\Omega_B\left(E - E_A, x - x_A\right)\right] = \ln\left[\Omega_B\left(E, x - x_A\right)\right] - \beta E_A + \mathcal{O}\left(E_A^2\right) \end{align} \]
There is
\[ \begin{align} \beta = \frac{1}{k_BT_K} \end{align} \]
is to be formed with the temperature of the heat bath, which, however, is equal to the temperature $T$ of the entire system, since the latter is in equilibrium (see Eq. (5.52)). Neglecting the higher-order terms, one obtains:
\[ \begin{align} P_r = \frac{\Omega_B\left(E, x - x_A\right)}{\Omega\left(E, x\right)}e^{-\beta E_r} = \frac{1}{Z}e^{-\beta E_r}. \end{align} \]
$Z = \frac{\Omega\left(E, x - x_A\right)}{\Omega_B\left(E, x\right)}$ is the canonical partition function; it acts as a normalization constant and ensures
\[ \begin{align} \sum_{r}^{}P_r = 1 \end{align} \]
:
In the grand canonical ensemble, not only heat but also particle exchange with the system $B$ is possible. Then, additionally, one has
\[ \begin{align} N = N_A + N_B = \text{const}. \end{align} \]
for the particle number. For the number of states $n\left(E_A, N_A\right)$ in which the system $A$ has energy $E_A$ and particle number $N_A$, one has
\[ \begin{align} n\left(E_A, N_A\right) = \Omega_A\left(E_A, N_A, x_A\right)\Omega_B\left(E_B, N_B, x_B\right). \end{align} \]
The probability $P\left(E_A, N_A\right)$ of finding a state $r$ with the energy $E_r = E_A$ and the particle number $N_r = N_A$ in the system $A$ is given by the probability
\[ \begin{align} P\left(E_A, N_A\right) &= \frac{n\left(E_A, N_A\right)}{\Omega\left(E, x\right)} = \frac{\Omega_A\left(E_A, N_A, x_A\right)\Omega_B\left(E_B, N_B, x_B\right)}{\Omega\left(E, x\right)}, \end{align} \]
to find the system $A$ with an energy $E_A$ and particle number $N_A$, divided by the number of these equally probable states:
\[ \begin{align} P_r = \frac{P\left(E_A, N_A\right)}{\Omega_A\left(E_A, N_A, x_A\right)} = \frac{\Omega_B\left(E - E_A, N - N_A, x_B\right)}{\Omega\left(E, x\right)} \end{align} \]
$E_B = E - E_A$, $N_B = N - N_A$ and $x_B = x - x_A$ were inserted. Because of $E_B\gg E_A$ and $N_B\gg N_A$, one can again expand the numerator:
\[ \begin{align} \ln\left[\Omega_B\left(E - E_A, N - N_A, x - x_A\right)\right] &= \ln\left[\Omega_B\left(E, N, x - x_A\right)\right] - \beta E_A - \frac{1}{k_B}\frac{\partial S}{\partial N}N_A\nonumber\\ & + \mathcal{O}\left(E_A^2, N_A^2, E_AN_A\right). \end{align} \]
The negative generalized force of the particle number is defined by the chemical potential
\[ \begin{align} \mu \coloneqq -\frac{\partial E}{\partial N} = -T\frac{\partial S\left(E, N.x\right)}{\partial N}. \end{align} \]
Neglecting the higher-order terms, one therefore has:
\[ \begin{align} P_r = \frac{\Omega_B\left(E, N, x - x_A\right)}{\Omega\left(E, x\right)}e^{-\beta\left(E_r - \mu N_r\right)} = \frac{1}{Y}e^{-\beta\left(E_r - \mu N_r\right)} \end{align} \]
$Y = \frac{\Omega\left(E, x\right)}{\Omega_B\left(E, N, x - x_A\right)}$ is the grand canonical partition function; it acts as a normalization constant and ensures
\[ \begin{align} \sum_{r}^{}P_r = 1 \end{align} \]
:
Now the first law is to be generalized to the case $dN \not= 0$. With Eqs. (5.29) and (5.32), it follows
\[ \begin{align} dE = dQ_{\text{qs}} - dW_{\text{qs}} = dQ_{\text{qs}} - \sum_{i}X_idx_i = dQ_{\text{qs}} - \frac{\partial E}{\partial V}dV - \frac{\partial E}{\partial N}dN = dQ_{\text{qs}} - pdV + \mu dN.\tag{5.82}\label{eq:td_1_id_gas} \end{align} \]
5.2.1 Thermodynamic potentials
A thermodynamic potential is a state variable from which all state variables can be determined by partial differentiation. In this section, only the volume $V$ and the particle number $N$ are considered as external parameters. In addition to the energy, the following thermodynamic potentials are defined:
\[ \begin{align} F \coloneqq E - ST& \text{(Helmholtz free energy)}\\ H \coloneqq E + pV& \text{(enthalpy)}\tag{5.84}\label{eq:def_enthalpie}\\ G \coloneqq E - ST + pV& \text{(Gibbs free energy)}\tag{5.85}\label{eq:def_gibbs-potential}\\ J \coloneqq E - ST - \mu N& \text{(grand canonical potential)}\tag{5.86}\label{eq:groskan_pot} \end{align} \]
For the differentials, with $dE = dQ - pdV + \mu dN$, one obtains
\[ \begin{align} dF &= -SdT - pdV + \mu dN, \tag{5.87}\label{eq:diff_free_energie}\\ dH &= TdS + Vdp + \mu dN,\\ dG &= -SdT + Vdp + \mu dN,\\ dJ &= -SdT - pdV - Nd\mu. \end{align} \]
The following statements can be read off from this:
\[ \begin{align} \left(\frac{\partial E}{\partial S}\right)_{V, N} = T, & {} & \left(\frac{\partial E}{\partial V}\right)_{S, N} = -p, & {} & \left(\frac{\partial E}{\partial N}\right)_{S, V} &= \mu\tag{5.91}\label{eq:chemisches_potential_prop_0}\\ \end{align} \] \[ \begin{align} \left(\frac{\partial F}{\partial T}\right)_{V, N} = -S, & {} & \left(\frac{\partial F}{\partial V}\right)_{T, N} = -p, & {} & \left(\frac{\partial F}{\partial N}\right)_{T, V} = \mu \end{align} \] \[ \begin{align} \left(\frac{\partial H}{\partial S}\right)_{p, N} = T, & {} & \left(\frac{\partial H}{\partial p}\right)_{S, N} = V, & {} & \left(\frac{\partial H}{\partial N}\right)_{S, p} = \mu \end{align} \] \[ \begin{align} \left(\frac{\partial G}{\partial T}\right)_{p, N} = -S, & {} & \left(\frac{\partial G}{\partial p}\right)_{T, N} = V, & {} & \left(\frac{\partial G}{\partial N}\right)_{T, p} = \mu\tag{5.95}\label{eq:chemisches_potential_prop_2} \end{align} \] \[ \begin{align} \left(\frac{\partial J}{\partial T}\right)_{V, \mu} = -S, & {} & \left(\frac{\partial J}{\partial V}\right)_{T, \mu} = -p, & {} & \left(\frac{\partial J}{\partial\mu}\right)_{T, V} = -N \end{align} \]
These are three statements each for the partial derivatives of $E$, $F$, $H$, $G$ and $J$. By applying the equality of the second partial derivatives, 15 non-trivial thermodynamic relations can be derived, which are called Maxwell relations:
\[ \begin{align} \left(\frac{\partial^2E}{\partial V\partial S}\right)_N = \left(\frac{\partial^2E}{\partial S\partial V}\right)_N&\Rightarrow\left(\frac{\partial T}{\partial V}\right)_{S, N} = -\left(\frac{\partial p}{\partial S}\right)_{V, N}\\ \left(\frac{\partial^2E}{\partial N\partial S}\right)_V = \left(\frac{\partial^2E}{\partial S\partial N}\right)_V&\Rightarrow\left(\frac{\partial T}{\partial N}\right)_{S, V} = \left(\frac{\partial \mu}{\partial S}\right)_{N, V} \end{align} \] \[ \begin{align} \left(\frac{\partial^2E}{\partial N\partial V}\right)_S = \left(\frac{\partial^2E}{\partial V\partial N}\right)_S&\Rightarrow -\left(\frac{\partial p}{\partial N}\right)_{V, S} = \left(\frac{\partial \mu}{\partial V}\right)_{N, S}\\ \left(\frac{\partial^2F}{\partial V\partial T}\right)_N = \left(\frac{\partial^2F}{\partial T\partial V}\right)_N&\Rightarrow\left(\frac{\partial S}{\partial V}\right)_{T, N} = \left(\frac{\partial p}{\partial T}\right)_{V, N} \end{align} \] \[ \begin{align} \left(\frac{\partial^2F}{\partial N\partial T}\right)_V = \left(\frac{\partial^2F}{\partial T\partial N}\right)_V&\Rightarrow -\left(\frac{\partial S}{\partial N}\right)_{T, V} = \left(\frac{\partial\mu}{\partial T}\right)_{N, V}\tag{5.101}\label{eq:maxwell_rel_4}\\ \left(\frac{\partial^2F}{\partial N\partial V}\right)_T = \left(\frac{\partial^2F}{\partial V\partial N}\right)_T&\Rightarrow -\left(\frac{\partial p}{\partial N}\right)_{V, T} = \left(\frac{\partial\mu}{\partial V}\right)_{N, T} \end{align} \] \[ \begin{align} \left(\frac{\partial^2H}{\partial p\partial S}\right)_N = \left(\frac{\partial^2H}{\partial S\partial p}\right)_N&\Rightarrow\left(\frac{\partial T}{\partial p}\right)_{S, N} = \left(\frac{\partial V}{\partial S}\right)_{p, N}\\ \left(\frac{\partial^2H}{\partial N\partial S}\right)_p = \left(\frac{\partial^2H}{\partial S\partial N}\right)_p&\Rightarrow\left(\frac{\partial T}{\partial N}\right)_{S, p} = \left(\frac{\partial\mu}{\partial S}\right)_{N, p} \end{align} \] \[ \begin{align} \left(\frac{\partial^2H}{\partial N\partial p}\right)_S = \left(\frac{\partial^2H}{\partial p\partial N}\right)_S&\Rightarrow\frac{V}{N} = \left(\frac{\partial V}{\partial N}\right)_{p, S} = \left(\frac{\partial\mu}{\partial p}\right)_{N, S}\tag{5.105}\label{eq:maxwell_rel_1}\\ \left(\frac{\partial^2G}{\partial p\partial T}\right)_N = \left(\frac{\partial^2G}{\partial T\partial p}\right)_N&\Rightarrow -\left(\frac{\partial S}{\partial p}\right)_{T, N} = \left(\frac{\partial V}{\partial T}\right)_{p, N} \end{align} \] \[ \begin{align} \left(\frac{\partial^2G}{\partial N\partial T}\right)_p = \left(\frac{\partial^2G}{\partial T\partial N}\right)_p&\Rightarrow -\frac{S}{N} = -\left(\frac{\partial S}{\partial N}\right)_{T, p} = \left(\frac{\partial\mu}{\partial T}\right)_{N, p}\tag{5.107}\label{eq:maxwell_rel_2}\\ \left(\frac{\partial^2G}{\partial N\partial p}\right)_T = \left(\frac{\partial^2G}{\partial p\partial N}\right)_T&\Rightarrow\left(\frac{\partial V}{\partial N}\right)_{p, T} = \left(\frac{\partial\mu}{\partial p}\right)_{N, T}\tag{5.108}\label{eq:maxwell_rel_3} \end{align} \] \[ \begin{align} \left(\frac{\partial^2J}{\partial T\partial V}\right)_\mu = \left(\frac{\partial^2J}{\partial V\partial T}\right)_\mu&\Rightarrow\left(\frac{\partial p}{\partial T}\right)_{\mu, V} = \left(\frac{\partial S}{\partial V}\right)_{\mu, T}\\ \left(\frac{\partial^2J}{\partial T\partial \mu}\right)_V = \left(\frac{\partial^2J}{\partial \mu\partial T}\right)_V&\Rightarrow\left(\frac{\partial N}{\partial T}\right)_{\mu, V} = \left(\frac{\partial S}{\partial\mu}\right)_{T, V} \end{align} \] \[ \begin{align} \left(\frac{\partial^2J}{\partial V\partial\mu}\right)_T = \left(\frac{\partial^2J}{\partial\mu\partial V}\right)_T&\Rightarrow\left(\frac{\partial N}{\partial V}\right)_{\mu, T} = \left(\frac{\partial p}{\partial\mu}\right)_{T, V}\tag{5.111}\label{eq:maxwell_rel_0} \end{align} \]
If one knows the microscopic structure of a system, one can determine the entropy, the microcanonical partition function, or the grand canonical partition function, depending on the physical requirements. The question is how one can determine macroscopic quantities such as compressibilities or heat capacities from the partition sums. To do this, one must be able to express one of the thermodynamic potentials in terms of the partition function of the system. First, the canonical ensemble is considered. In this case $\left(T, V, N\right)$ or alternatively $\left(\beta, V, N\right)$ are fixed. One has
\[ \begin{align} d\ln\left[Z\left(\beta, V, N\right)\right] = \frac{\partial\ln\left(Z\right)}{\partial\beta}d\beta + \frac{\partial\ln\left(Z\right)}{\partial V}dV + \frac{\partial\ln\left(Z\right)}{\partial N}dN. \end{align} \]
Now the partial derivatives of the partition function Eq. (5.72) calculated:
\[ \begin{align} \frac{\partial\ln\left(Z\right)}{\partial\beta} &= -\frac{1}{Z}\sum_{r}^{}E_r\exp\left(-\beta E_r\right) = -E\\ \frac{\partial\ln\left(Z\right)}{\partial V} &= -\frac{\beta}{Z}\sum_r\frac{\partial E_r\left(V, N\right)}{\partial V}\exp\left(-\beta E_r\right) = -\beta\newoverline{\frac{\partial E_r}{\partial V}} = \beta p \end{align} \] \[ \begin{align} \frac{\partial\ln\left(Z\right)}{\partial N} &= -\frac{\beta}{Z}\sum_r\frac{\partial E_r\left(V, N\right)}{\partial N}\exp\left(-\beta E_r\right) = -\beta\newoverline{\frac{\partial E_r}{\partial N}} = -\beta\mu \end{align} \]
Thus one has
\[ \begin{align} d\ln\left(Z\left(\beta, V, N\right)\right) &= -Ed\beta + \beta pdV - \beta\mu dN\nonumber\\ \Rightarrow d\left(\ln\left(Z\right) + \beta E\right) &= \beta\left(dE + pdV - \mu dN\right) = \frac{1}{k_B}\left(\frac{dE}{T} + \frac{p}{T}dV - \frac{\mu}{T}dN\right) \end{align} \]
By comparison with Eq. (5.47), one obtains
\[ \begin{align} k_Bd\left[\ln\left(Z\right) + \beta E\right] &= dS\Rightarrow S = k_B\left[\ln\left(Z\right) + \beta E\right] + C. \end{align} \]
The constant $C$ turns out to be zero, which will not be shown here; thus it follows
In the grand canonical ensemble, $\left(T, V, \mu\right)$ are fixed; one obtains the differential
\[ \begin{align} d\ln\left[Y\left(\beta, V, \mu\right)\right] = \frac{\partial\ln\left(Y\right)}{\partial\beta}d\beta + \frac{\partial\ln\left(Y\right)}{\partial V}dV + \frac{\partial\ln\left(Y\right)}{\partial\mu}d\mu. \end{align} \]
The partial derivatives are calculated using the partition function Eq. (5.81)
\[ \begin{align} \frac{\partial\ln\left(Y\right)}{\partial\beta} &= -\frac{1}{Y}\sum_{r}^{}\left(E_r - \mu N_r\right)\exp\left(-\beta \left(E_r - \mu N_r\right)\right) = -E + \mu N,\\ \frac{\partial\ln\left(Y\right)}{\partial V} &= -\frac{\beta}{Y}\sum_r\frac{\partial E_r\left(V, N\right)}{\partial V}\exp\left(-\beta \left(E_r - \mu N_r\right)\right) = -\beta\newoverline{\frac{\partial E_r}{\partial V}} = \beta p, \end{align} \] \[ \begin{align} \frac{\partial\ln\left(Y\right)}{\partial \mu} &= \frac{\beta}{Y}\sum_r N_r\exp\left(-\beta \left(E_r - \mu N_r\right)\right) = \beta\newoverline{N_r} = \beta N. \end{align} \]
Thus one has
\[ \begin{align} d\ln\left[Y\left(\beta, V, \mu\right)\right] &= \left(-E + \mu N\right)d\beta + \beta pdV + \beta Nd\mu\nonumber\\ \Leftrightarrow d\left[\ln\left(Y\right) + \beta E - \beta\mu N\right] &= \beta\left(dE + pdV - \mu dN\right) = \frac{dS}{k_B}. \end{align} \]
With this, one has
\[ \begin{align} S = k_B\left[\ln\left(Y\right) + \beta E - \beta\mu N\right]\Leftrightarrow k_BT\ln\left(Y\right) = TS - E + \mu N. \end{align} \]
A possible constant here is also zero. By comparison with Eq. (5.86), it follows
The equations (5.118) and (5.125) provide a connection between microscopic statistics and macroscopic thermodynamics.
For the total differential $d\mu$ of the chemical potential $\mu = \mu\left(T, p\right)$, one has
\[ \begin{align} d\mu = \left(\frac{\partial\mu}{\partial T}\right)_pdT + \left(\frac{\partial\mu}{\partial p}\right)_Tdp \stackrel{\href{#eq:maxwell_rel_4}{\text{Eqs. (5.101), (5.108)}}}{=} -\left(\frac{\partial S}{\partial N}\right)_{T, V}dT + \left(\frac{\partial V}{\partial N}\right)_{p, T}dp. \end{align} \]
With
\[ \begin{align} \left(\frac{\partial S}{\partial N}\right)_{T, V} = \frac{S}{N} =: s, & {} & \left(\frac{\partial V}{\partial N}\right)_{p, T} = \frac{V}{N} =: v \end{align} \]
it follows
which is called the Gibbs-Duhem relation.
5.3 Ideal gas
An ideal gas is a gas in which the particles do not interact with each other. The Hamiltonian of an ideal gas made of $N$ particles is
\[ \begin{align} \newhat{H} = \sum_{i = 1}^{N} - \frac{\hbar^2}{2m}\Delta_i + V\left(\mathbf{r}_i\right) = \sum_{i = 1}^{N}\newhat{H}_i, \end{align} \]
it is a superposition of one-particle Hamiltonians
\[ \begin{align} \newhat{H}_i = -\frac{\hbar^2}{2m}\Delta_i + V\left(\mathbf{r}_i\right). \end{align} \]
$V\left(\mathbf{r}_i\right)$ is the familiar wall potential
\[ \begin{align} V\left(\mathbf{r}_i\right) = \begin{cases} 0, \:\mathbf{r}_i\in V,\\ \infty, \text{ otherwise}, \end{cases} \end{align} \]
where $V$ is the volume to which the particles are confined. Here $V = L^3$ is a cube with edge length $L$. This corresponds to a 3D potential well, see Sect. 4.2. The one-particle solution is
\[ \begin{align} \psi\left(\mathbf{r}_i\right)\propto\sin\left(k_{3i - 2}x\right)\sin\left(k_{3i - 1}y\right)\sin\left(k_{3i}z\right). \end{align} \]
Here, the boundary condition holds for $k_{3i - 2}, k_{3i - 1}, k_{3i}$
\[ \begin{align} \frac{Lp_i}{\hbar} = k_iL = n_i\pi\tag{5.133}\label{eq:deriv_ideal_gas_bc} \end{align} \]
with $n_i\in \mathbb {N}$ with $n\geq 1$. $p_i = \hbar k_i$ is the momentum. The particles do not penetrate the wall, so they have no potential energy. Thus, for the total energy of the system, one has
\[ \begin{align} E = \frac{1}{2m}\sum_{i = 1}^{3N}p_i^2. \end{align} \]
To determine the microcanonical partition function of the ideal gas, one starts from $\delta E = E$:
\[ \begin{align} \Omega\left(E, V, N\right) &= \sum_{E_r<E}1 = \underbrace{\sum_{n_1 = 1, 2, \dotsc}\dotsc\sum_{n_{3N} = 1, 2, \dotsc}}_{E_r < E} 1 \approx \frac{1}{2^{3N}}\underbrace{\int\dotsc\int}_{E_r < E}dn_1\dotsc dn_{3N}. \end{align} \]
Because $\newoverline{n_i}\gg 1$, the sums were replaced by integrals in the last step. The factor $1/2^{3N}$ arises from the inclusion of negative values in the integration (there are $3N$ coordinate axes that must be halved). Eq. (5.133) implies
\[ \begin{align} dn_i = \frac{L}{\hbar\pi}dp_i. \end{align} \]
From this it follows
\[ \begin{align} \Omega\left(E, V, N\right) &= \left(\frac{L}{2\hbar\pi}\right)^{3N}\underbrace{\int\dotsc\int}_{\sum p_i^2< 2mE}dp_1\dotsc dp_{3N} = \frac{V^N}{\left(2\hbar\pi\right)^{3N}}\underbrace{\int\dotsc\int}_{\sum p_i^2< 2mE}dp_1\dotsc dp_{3N}. \end{align} \]
The remaining 3N-dimensional integral is the volume of a 3N-dimensional sphere of radius $\sqrt{2mE}$, so Eq. (B.143) can be used:
\[ \begin{align} \Omega\left(E, V, N\right) &= \frac{V^N}{\left(2\hbar\pi\right)^{3N}}\frac{\pi^{3N/2}}{\Gamma\left(\frac{3N}{2} + 1\right)}\sqrt{2mE}^{3N} \stackrel{\href{ch-39-derivations-of-some-mathematical-formula.html#eq:gamma_prop_1}{\text{Eq. (A.113)}}}{=} \frac{V^N}{\left(2\pi\hbar\right)^{3N}}\frac{\pi^{3N/2}}{\left(3N/2\right)!}\left(2m\right)^{3N/2}E^{3N/2}\nonumber\\ &= \frac{V^N}{\left(2\pi\hbar\right)^{3N}}\frac{1}{\left(3N/2\right)!}\left(2m\pi\right)^{3N/2}E^{3N/2} = \left[\frac{2\pi m}{\left(2\pi\hbar\right)^2}\right]^{3N/2}\frac{E^{3N/2}}{\left(3N/2\right)!}V^N \end{align} \]
Using the Stirling formula, Eq. (A.119), one obtains
\[ \begin{align} \Omega\left(E, V, N\right) &= \left[\frac{2\pi m}{\left(2\pi\hbar\right)^2}\right]^{3N/2}E^{3N/2}V^N\frac{1}{\left(3N/2\right)!} \approx \sqrt{\frac{1}{2\pi}}\left[\frac{2\pi m}{\left(2\pi\hbar\right)^2}\right]^{3N/2}e^{3N/2}\left(\frac{2}{3N}\right)^{\frac{3N}{2} + \frac{1}{2}}E^{3N/2}V^N\nonumber\\ &= \frac{1}{\sqrt{3N\pi}}\left[\frac{2\pi me}{\left(2\pi\hbar\right)^2}\right]^{3N/2}\left(\frac{2}{3N}\right)^{\frac{3N}{2}}E^{3N/2}V^N = \frac{1}{\sqrt{3\pi N}}\left[\frac{4\pi me}{3\left(2\pi\hbar\right)^2}\right]^{3N/2}V^N\left(\frac{E}{N}\right)^{3N/2}. \end{align} \]
This result is not yet correct; an important modification still needs to be made. The $N$ particles are quantum mechanically indistinguishable, which means that swapping two particles changes nothing. So $N!$ states are the same, which is why one must make the replacement
\[ \begin{align} \Omega\to\frac{\Omega}{N!} \end{align} \]
From this it follows, again using the Stirling formula,
\[ \begin{align} \Omega\left(E, V, N\right) &= \frac{1}{\sqrt{6\pi^2}}\frac{1}{N}\left[\frac{4\pi me^{5/3}}{3\left(2\pi\hbar\right)^2}\right]^{3N/2}\left(\frac{V}{N}\right)^N\left(\frac{E}{N}\right)^{3N/2}\nonumber\\ &= \frac{1}{\sqrt{6\pi^2}}\frac{1}{N}\left[\frac{me^{5/3}}{3\pi\hbar^2}\right]^{3N/2}\left(\frac{V}{N}\right)^N\left(\frac{E}{N}\right)^{3N/2}. \end{align} \]
For the entropy, it follows
\[ \begin{align} \frac{S\left(E, V, N\right)}{k_B} &= \frac{1}{2}\ln\left(\frac{1}{6\pi^2}\right) - \ln\left(N\right) + \frac{3N}{2}\ln\left(c\right) + N\ln\left(\frac{V}{N}\right) + \frac{3N}{2}\ln\left(\frac{E}{N}\right)\nonumber\\ &\approx \frac{3N}{2}\ln\left(c\right) + N\ln\left(\frac{V}{N}\right) + \frac{3N}{2}\ln\left(\frac{E}{N}\right) - \ln\left(N\right)\nonumber\\ &\approx \frac{3N}{2}\ln\left(c\right) + N\ln\left(\frac{V}{N}\right) + \frac{3N}{2}\ln\left(\frac{E}{N}\right)\nonumber \end{align} \]
with
\[ \begin{align} c \coloneqq \frac{me^{5/3}}{3\pi\hbar^2}.\tag{5.143}\label{eq:def_id_gas_entropy_constant} \end{align} \]
Eq. (5.142) is also called the Sackur-Tetrode equation. One obtains the caloric equation of state of ideal gases
\[ \begin{align} \frac{1}{k_BT} = \frac{\partial\ln\left(\Omega\left(E, V, N\right)\right)}{\partial E} = \frac{3}{2}\frac{N}{E}\Rightarrow E = \frac{3}{2}Nk_BT.\tag{5.144}\label{eq:kalorisch_id_gase} \end{align} \]
Furthermore, one has
\[ \begin{align} \frac{p}{T} = \frac{\partial S\left(E, V, N\right)}{\partial V} = k_BN\frac{1}{V}. \end{align} \]
This gives the thermal equation of state of ideal gases
For the isochoric heat capacity of the ideal gas, one obtains with Eq. (5.144)
\[ \begin{align} C^{(V)} &= \frac{3}{2}k_BN. \end{align} \]
For the corresponding specific quantity, one has
\[ \begin{align} c^{(V)} &= \frac{\frac{3}{2}k_BN}{Nm} = \frac{3k_B}{2m}.\tag{5.148}\label{eq:c_v_ideal_gas} \end{align} \]
Thus, for the energy $E$ one can write
\[ \begin{align} E = C^{(V)}T = Nmc^{(V)}T\tag{5.149}\label{eq:kalorisch_id_gase_mod} \end{align} \]
For the isobaric heat capacity, one has with Eq. (5.60)
\[ \begin{align} C^{(p)} = T\left(\frac{\partial S}{\partial T}\right)_{p} = k_BTN\frac{\partial}{\partial T}\left(\ln\left(\frac{k_BT}{p}\right) + \frac{3}{2}\ln\left(\frac{3}{2}Nk_BT\right)\right) = k_BTN\left(\frac{1}{T} + \frac{3}{2}\frac{1}{T}\right) = \frac{5}{2}Nk_B. \end{align} \]
For the enthalpy $H$ of the ideal gas, it follows
\[ \begin{align} H = E + pV = \frac{5}{2}Nk_BT = C^{(p)}T. \end{align} \]
For the Gibbs potential $G$ of the ideal gas one obtains
\[ \begin{align} G = E - ST + pV = H - ST = C^{(p)}T - ST\tag{5.152}\label{eq:gibbs_ideal}. \end{align} \]
Furthermore, one has
\[ \begin{align} c^{(p)} - c^{(V)} = \frac{N}{m}k_B = \frac{N}{m N_A}R = R_s\tag{5.153}\label{eq:diff_spez_heat_id_gase}. \end{align} \]
For the chemical potential, one has with Eqs. (5.85) and (5.95)
\[ \begin{align} \mu = \frac{E - TS + pV}{N} = \frac{C^{(v)}T}{N} - \frac{ST}{N} + k_BT = T\left(\frac{C^{(v)}}{N} - \frac{S}{N} + k_B\right),\tag{5.154}\label{eq:chemical_potential_id_gas} \end{align} \]
where it was used that the Gibbs potential is an extensive quantity. An equivalent formulation of Eq. (5.146) is
\[ \begin{align} p = \frac{Nk_BT}{V} = \frac{nN_Ak_BT}{V} = \frac{nRT}{V} = \frac{mRT}{VM} = \rho R_sT\tag{5.155}\label{eq:zustand_ideal_alt} \end{align} \]
with the individual gas constant $R_s \coloneqq \frac{R}{M}$.
The equation of state applies to a gas, not to a gas mixture. However, air consists of various gases. Now the equation of state is to be extended to such a mixture. So let a gas with $N \in \mathbb{N}$ components be given. For $1 \le i \le N$ let $p_i$ be the partial pressure of the $i-$th gas component. Since there are no interactions between the components, the following then holds:
\[ \begin{align} p = \sum\limits_{i = 1}^{N}p_i = \sum\limits_{i = 1}^{N}\rho_iR_iT = \rho T\sum\limits_{i = 1}^{N}R_i\frac{\rho_i}{\rho} = TR\rho \sum\limits_{i = 1}^{N}\frac{m_i}{m}\frac{1}{M_i}. \end{align} \]
With the definition
\[ \begin{align} M \coloneqq \frac{1}{\sum\limits_{i = 1}^{N}\frac{m_i}{m}\frac{1}{M_i}}\tag{5.157}\label{eq:mittlere_molare_masse} \end{align} \]
one obtains
\[ \begin{align} p = \rho\frac{R}{M}T = \rho R_s T \end{align} \]
also for a gas mixture. The average molar mass according to Eq. (5.157) can be easily calculated by weighting the molar masses of the components with their volume fractions:
\[ \begin{align} M = \frac{1}{\sum_{i = 1}^{N}\frac{m_i}{m}\frac{n_i}{m_i}} = \frac{\sum_{i = 1}^{N}n_iM_i}{\sum_{i = 1}^{N}n_i} = \sum_{i = 1}^{N}\frac{n_i}{n}M_i, \end{align} \]
Here $n_i$ is the amount of substance of the $i-$th component and $n$ the total amount of substance. In terms of the mass densities, one can write
\[ \begin{align} M &= \frac{m}{n} = \frac{m}{\sum_i\frac{m_i}{M_i}} = \frac{\rho}{\sum_i\frac{\rho_i}{M_i}} = \frac{1}{\frac{1}{\rho}\sum_i\frac{\rho_i}{M_i}}. \end{align} \]
For the molar mass of moist air $M_h$, one has
\[ \begin{align} M_h &= \frac{1}{\frac{\rho_d}{M_d\rho_h} + \frac{\rho_v}{\rho_h}\frac{1}{M_v}}. \end{align} \]
Thus one has
\[ \begin{align} \frac{1}{M_h} = \frac{1}{M_d}\left(1 - \frac{\rho_v}{\rho_h}\right) + \frac{\rho_v}{\rho_h}\frac{1}{M_v}. \end{align} \]
The individual gas constant of moist air is therefore
\[ \begin{align} R_h = R_d\left(1 - \frac{\rho_v}{\rho_h} + \frac{\rho_v}{\rho_h}\frac{M_d}{M_v}\right).\tag{5.163}\label{eq:gaskonstanth_humider_luft} \end{align} \]
One further defines the virtual temperature $T_v$ as the temperature at which dry air would have the same density as moist air at the same pressure. As an equation this means
\[ \begin{align} R_h\rho T &= R_d\rho T_v\Rightarrow T_v = \frac{R_h}{R_d}T = T\left(1 - \frac{\rho_v}{\rho_h} + \frac{\rho_v}{\rho_h}\frac{M_d}{M_v}\right)\nonumber\\ &= T\left[1 + \frac{\rho_v}{\rho_h}\left(\frac{M_d}{M_v} - 1\right)\right].\tag{5.164}\label{eq:def_virtual_temperature} \end{align} \]
The difference
\[ \begin{align} \Delta T_v& \coloneqq T_v - T = T\frac{\rho_v}{\rho_h}\left(\frac{M_d}{M_v} - 1\right) \end{align} \]
is called the virtual temperature surcharge. The decisive advantage of the virtual temperature is that the spatial dependence of the individual gas constant $R_h$ is absorbed into the temperature, which is particularly convenient for spatial and temporal derivatives. Therefore, virtual temperature plays an important role in modeling.
Now the humidity measures are to be discussed. The density $\rho_v$ (condensation products are neglected here) is also referred to as absolute humidity and the vapor pressure has already been introduced. First the relative humidity $U$ is given by
\[ \begin{align} U \coloneqq\frac{p_v}{p_v^{(S)}} \end{align} \]
with $p_v$ as the partial pressure of water vapor and $p_v^{(S)}$ as the saturation vapor pressure. One further defines the specific humidity $q$ by
\[ \begin{align} q \coloneqq\frac{\rho_v}{\rho}\tag{5.167}\label{eq:def_spec_humidity} \end{align} \]
The mixing ratio $r$ is defined by
\[ \begin{align} r \coloneqq\frac{\rho_v}{\rho_d} \end{align} \]
Furthermore, the volume mixing ratio $r_V$ is given by
\[ \begin{align} r_V \coloneqq\frac{n_v}{n_d} \end{align} \]
defined. This quantity is often used in chemical and radiation considerations where particle density rather than mass density is important. A superscript $S$ on the symbols means that saturation is assumed. Some useful conversions are
\[ \begin{align} q &= \frac{p_vR_hT}{R_vT p} = \frac{p_vR_h}{pR_v} = \frac{p_v}{p}\frac{R_d}{R_v}\left(1 - \frac{\rho_v}{\rho_h} + \frac{\rho_v}{\rho_h}\frac{M_d}{M_v}\right)\nonumber\\ &= \frac{p_v}{p}\left(\frac{M_v}{M_d}\left(1 - q\right) + q\right) = \frac{p_v}{p}\left(q\left(1 - \frac{M_v}{M_d}\right) + \frac{M_v}{M_d}\right)\nonumber\\ \Rightarrow q\left(1 + \left(\frac{M_v}{M_d} - 1\right)\frac{p_v}{p}\right) &= \frac{M_v}{M_d}\frac{p_v}{p}\Rightarrow q = \frac{\frac{M_v}{M_d}}{\frac{p}{p_v} + \frac{M_v}{M_d} - 1},\\ r &= \frac{\rho_v}{\rho_d} = \frac{p_vR_dT}{R_vTp_d} = \frac{p_v}{p_d}\frac{M_v}{M_d} = \frac{p_v}{p - p_v}\frac{M_v}{M_d} = \frac{\frac{M_v}{M_d}}{\frac{p}{p_v} - 1}, \nonumber\\ r_V &= \frac{n_v}{n_d} = \frac{p_v}{p_d} = \frac{\rho_vR_v}{\rho_dR_d}. \end{align} \]
Common approximations are
\[ \begin{align} q \approx \frac{\frac{M_v}{M_d}}{\frac{p}{p_v} - 1} = r, & {} & q \approx \frac{M_v}{M_d}\frac{p_v}{p}, & {} & r \approx \frac{M_v}{M_d}\frac{p_v}{p}.\tag{5.172}\label{eq:mischungsverhaeltnis_vereinfacht} \end{align} \]
5.4 Classic systems
Classical phase space coordinates are continuous and therefore difficult to grasp statistically. Semi-classical derivations are easier, in which one starts from the QM and then carries out the classical transition $\hbar\to0$.
Let $N$ particles be enclosed in a cubic volume $V = L^3$. For every wave-vector component of the one-particle wave functions, one has
\[ \begin{align} k_nL = n\pi\Leftrightarrow k_n = \frac{n\pi}{L} \end{align} \]
with $n\in\mathbb{N}$ and $n\geq 1$. Integrating over the entire $k$-space, one can replace the sums by integrals:
\[ \begin{align} \sum_{\mathbf{k}}\dotsc = \frac{V^N}{\left(2\pi\right)^{3N}}\int_{}\dotsc\int_{}\dotsc dk_1\dotsc dk_{3N} \end{align} \]
The factor $1/2$ before each integral is obtained by including negative $k$ values in the integration. In momentum space one obtains
\[ \begin{align} \sum_{\mathbf{k}}\dotsc = \frac{V^N}{\left(2\pi\hbar\right)^{3N}}\int\dotsc d^{3N}p. \end{align} \]
Now this is to be generalized to phase space. Consider a particle in a 1D potential well $0\leq x\leq L$ with $L>0$; there exist solutions of the SE of the form
\[ \begin{align} \psi_n\left(x\right) = \sqrt{\frac{2}{L}}\sin\left(n\frac{\pi}{L}x\right), \end{align} \]
Here let $n\in \mathbb {N}$ with $n\geq 1$; the energy eigenvalues are given by
\[ \begin{align} E_n = \frac{\hbar^2n^2\pi^2}{2mL^2} \end{align} \]
For the phase space volume $V\left(E\right)$ of a state with energy $E$, one has
\[ \begin{align} V\left(E\right) = \int_{0}^{L}\int_{p^2/2m\leq E} dpdx = L2\sqrt{2mE}. \end{align} \]
For a given energy $E\gg 1$, the number of states $N\left(E\right)$ with eigenenergies $E_n\leq E$ is approximately given by
\[ \begin{align} N\left(E\right)\approx\frac{L}{\pi\hbar}\sqrt{2mE}. \end{align} \]
One thus obtains
\[ \begin{align} \frac{V\left(E\right)}{N\left(E\right)} = \frac{2L\sqrt{2mE}}{\sqrt{2mE}}\frac{\pi\hbar}{L} = 2\pi\hbar. \end{align} \]
This result applies generally, i.e. also for more complicated potentials. In the case of $f$ degrees of freedom, a microstate occupies the phase space volume $\left(2\pi\hbar\right)^f$. Summing over all microstates $r$ and again replacing this by an integral, one obtains
\[ \begin{align} \sum_{r}^{}\dotsc = \frac{1}{\left(2\pi\hbar\right)^f}\int\dotsc dxdp. \end{align} \]
An averaging operator results
\[ \begin{align} \sum_{r}^{}P_r\dotsc = \frac{1}{\left(2\pi\hbar\right)^f}\int\rho'\left(x, p\right)\dotsc dxdp\tag{5.182}\label{eq:mittelungsoperator_klassisch} \end{align} \]
with the probabilities $P_r$ of the microstates and a modified probability density $\rho'\left(x, p\right)$.
5.4.0.1 Maxwell distribution
Inserting the canonical distribution
\[ \begin{align} P_r = \frac{1}{Z}\exp\left(-\frac{E_r}{k_BT}\right) \end{align} \]
into Eq. (5.182), one obtains
\[ \begin{align} \frac{1}{Z}\sum_{r}^{}\exp\left(-\frac{E_r}{k_BT}\right)\dotsc &= \frac{1}{Z\left(2\pi\hbar\right)^f}\int\exp\left(-\frac{E\left(x, p\right)}{k_BT}\right)\dotsc dxdp \end{align} \]
If the integration depends only on the momentum, it follows
\[ \begin{align} \frac{1}{Z}\sum_{r}^{}\exp\left(-\frac{E_r}{k_BT}\right)\dotsc &= \frac{V}{Z\left(2\pi\hbar\right)^f}\int\exp\left(-\frac{E\left(p\right)}{k_BT}\right)\dotsc dp \end{align} \]
You interpret
\[ \begin{align} \rho\left(p\right) = \frac{V}{Z\left(2\pi\hbar\right)^f}\exp\left(-\frac{E\left(p\right)}{k_BT}\right) \end{align} \]
as a probability density. Now one considers exactly one particle and assumes Cartesian momenta, so that:
\[ \begin{align} \rho\left(p\right) = \frac{4\pi V}{z\left(2\pi\hbar\right)^3}\exp\left(-\frac{p^2}{2mk_BT}\right)p^2 \end{align} \]
For the canonical partition function $z$ of this single particle, it follows
\[ \begin{align} z\left(T, V\right) &= \frac{1}{\left(2\pi\hbar\right)^3}V\int_{0}^{\infty}4\pi\exp\left(-\frac{p^2}{2mk_BT}\right)p^2dp = \frac{V}{2\pi^2\hbar^3}\int_{0}^{\infty}\exp\left(-\frac{p^2}{2mk_BT}\right)p^2dp. \end{align} \]
With Eq. (A.109) follows
\[ \begin{align} z\left(T, V\right) &= \frac{V}{8\pi^2\hbar^3}\sqrt{\pi\left(2mk_BT\right)^3} = \frac{V}{\hbar^3}\left(\frac{mk_BT}{2\pi}\right)^{3/2}. \end{align} \]
From this it follows
\[ \begin{align} \rho\left(p\right) &= \frac{\hbar^3}{v}_h\left(\frac{2\pi}{mk_BT}\right)^{3/2}\frac{4\pi V}{\left(2\pi\hbar\right)^3}\exp\left(-\frac{p^2}{2mk_BT}\right)p^2 = \sqrt{\frac{2}{m^3\pi k_B^3T^3}}\exp\left(-\frac{p^2}{2mk_BT}\right)p^2. \end{align} \]
The velocity distribution therefore reads:
This is the Maxwell distribution. The maximum $v_{\mathrm{max}}$ is obtained from the condition
\[ \begin{align} 0 = 2ve^{-\frac{mv^2}{2k_BT}} - \frac{mv^3}{k_BT}e^{-\frac{mv^2}{2k_BT}} \Leftrightarrow v_{\text{max}} = \sqrt{\frac{2k_BT}{m}}. \end{align} \]
If one substitutes the molar mass of dry air and 20$^\circ$ C for $m$, one obtains $v_{\mathrm{max}} \approx 4\cdot 10^2$ m/s. The expectation value of this distribution is
\[ \begin{align} \newoverline{v} = \sqrt{\frac{2m^3}{\pi k_B^3T^3}}\int_{0}^{\infty}e^{-\frac{mv^2}{2k_BT}}v^3dv. \end{align} \]
With $C = \frac{m}{2k_BT}$ in Eq. (A.110), it follows
\[ \begin{align} \newoverline{v} = \sqrt{\frac{2m^3}{\pi k_B^3T^3}}\frac{2k_B^2T^2}{m^2} = \sqrt{\frac{8k_BT}{\pi m}}. \end{align} \]
The corresponding vectorial probability density $\rho\left(\mathbf{v}\right)$ is
\[ \begin{align} \rho\left(\mathbf{v}\right) = \left(\frac{m}{2\pi k_BT}\right)^{3/2}\exp\left(-\frac{mv^2}{2k_BT}\right).\tag{5.195}\label{eq:maxwellverteilung_vektoriell} \end{align} \]
Also important is the expectation value of the relative velocity of two particles of the same mass. For this, one has
\[ \begin{align} \mathbf{v}_{\text{rel}} = \mathbf{v}_2 - \mathbf{v}_1. \end{align} \]
For the probability density $\rho\left(\mathbf{v}_1, \mathbf{v}_2\right)$ of two particle velocities $\mathbf{v}_1, \mathbf{v}_2$, one has
\[ \begin{align} \rho\left(\mathbf{v}_1, \mathbf{v}_2\right) = \left(\frac{m}{2\pi k_BT}\right)^3\exp\left(-\frac{m\left(v_1^2 + v_2^2\right)}{2k_BT}\right). \end{align} \]
From this, with the abbreviations
\[ \begin{align} a \coloneqq \left(\frac{m}{2\pi k_BT}\right)^{3/2}, & {} & b \coloneqq \frac{m}{2k_BT} \end{align} \]
one obtains the relation
\[ \begin{align} \rho\left(\mathbf{v}_{\text{rel}}\right) &= a^2\int\exp\left[-b\left(v_1^2 + v_1^2 + v_{\text{rel}}^2 + 2v_{1, x}v_{rel, x} + 2v_{1, y}v_{rel, y} + 2v_{1, z}v_{rel, z}\right)\right]dv_1^3\nonumber\\ &= a^2e^{-bv_{\text{rel}}^2}\int\exp\left[-2b\left(v_1^2 + v_{1, x}v_{rel, x} + v_{1, y}v_{rel, y} + v_{1, z}v_{rel, z}\right)\right]dv_1^3\nonumber\\ &= a^2e^{-bv_{\text{rel}}^2}\int\exp\left[-2b\left(\left(v_{1, x} + \frac{v_{rel, x}}{2}\right)^2 + \dotsc - \frac{v_{\text{rel}}^2}{4}\right)\right]dv_1^3\nonumber\\ &= a^2e^{-\frac{b}{2}v_{\text{rel}}^2}\int e^{-2bv_1^2}dv_1^3 = ae^{-\frac{b}{2}v_{\text{rel}}^2}\int ae^{-b\left(\sqrt{2}v_1\right)^2}dv_1^3 = ae^{-\frac{b}{2}v_{\text{rel}}^2}\frac{1}{2^{3/2}}. \end{align} \]
The last step follows from the normalization of the distribution Eq. (5.195). Thus, for the probability density of the magnitude of the relative velocity, one has
\[ \begin{align} \rho\left(v_{\text{rel}}\right) &= \frac{4\pi a}{2^{3/2}}e^{-\frac{b}{2}v_{\text{rel}}^2}v_{\text{rel}}^2. \end{align} \]
For the expectation value, it follows
\[ \begin{align} \newoverline{v_{\text{rel}}} &= \int_{0}^{\infty}\frac{4\pi a}{2^{3/2}}e^{-\frac{b}{2}v_{\text{rel}}^2}v_{\text{rel}}^3dv_{\text{rel}} = \sqrt{2}\int_{0}^{\infty}4\pi ae^{-bv_{\text{rel}}^2}v_{\text{rel}}^3dv_{\text{rel}} = \sqrt{2}\newoverline{v}.\tag{5.201}\label{eq:mittlere_rel_gesch} \end{align} \]
5.4.0.2 Kinetic gas model
In a scattering experiment, a current density $j$ (incident particles per area and time) falls on a target. Multiplying the current density by an area gives a particle current. One defines the effective cross section $\sigma$ by
\[ \begin{align} j\sigma \coloneqq \frac{dN_{\text{str}}}{dt}, \end{align} \]
It therefore corresponds to the area on which particles are effectively scattered. This is now applied to a gas made of particles that are hard spheres of diameter $d$. In this case, one has
\[ \begin{align} \sigma = \pi d^2. \end{align} \]
A particle travels the path $l$, colliding with $N$ particles:
\[ \begin{align} N = n\sigma l \end{align} \]
Here $n$ is the particle density and $\sigma l$ is the volume of a cylinder with radius $d$ in which collision partners are struck. For the mean collision time $\tau$, one therefore has
\[ \begin{align} 1 &= n\sigma\newoverline{v_{\text{rel}}}\tau\nonumber \end{align} \]
For the mean free path $\lambda$ follows
Inhomogeneous properties of a gas are equalized through transport processes. This will be understood here using a simple model; the values determined for the transport constants should be understood as estimates. So let $q$ be any property of the gas. $q$ depends only on $z$, $q = q\left(z\right)$. The velocity distribution is isotropic, which is represented here by the fact that just $1/6$ of all particles fly in each Cartesian coordinate direction. In doing so, they transport their properties on average with a length $\lambda$. This gives the current density $j_z$ at $z = z_0$
\[ \begin{align} j_z = \frac{\text{transport}}{\text{time}\times\text{area}} = \frac{1}{6}\left(\left(n\newoverline{v}q\right)\left(z_0 - \lambda\right) - \left(n\newoverline{v}q\right)\left(z_0 + \lambda\right)\right)\approx\frac{1}{6}\frac{\partial\left(n\newoverline{v}q\right)}{\partial z}\left(-2\lambda\right) = -\frac{\lambda}{3}\frac{\partial\left(n\newoverline{v}q\right)}{\partial z}. \end{align} \]
This can be written as:
\[ \begin{align} j_z = -C\frac{\partial q}{\partial z},\tag{5.208}\label{eq:diffusionsstrom} \end{align} \]
if one assumes that $q$ is not correlated with other quantities. For $q = 1$ and homogeneous $v$ one obtains Diffusion,
\[ \begin{align} C\to D\approx\frac{\newoverline{v}\lambda}{3},\tag{5.209}\label{eq:diff_const_kinetic_gastheory} \end{align} \]
$D$ is the diffusion constant. With the continuity equation
\[ \begin{align} \frac{\partial n}{\partial t} = -\frac{\partial}{\partial z}\left(j_z\right) \end{align} \]
follows the diffusion equation
\[ \begin{align} \frac{\partial n}{\partial t} = D\frac{\partial^2n}{\partial z^2}. \end{align} \]
or in 3D
\[ \begin{align} \frac{\partial n}{\partial t} &= D\Delta n.\tag{5.212}\label{eq:diffusionsglg} \end{align} \]
If one sets $q = mu$ with $u$ as the flow velocity in the x-direction, one examines the momentum current density. $u$ is obtained by averaging the particle velocities, where $u\ll\newoverline{v}$ should apply.
\[ \begin{align} j_z &= \frac{\text{momentum}}{\text{time}\times\text{area}} = \frac{1}{6}\left(\left(n\newoverline{v}mu\right)\left(z_0 - \lambda\right) - \left(n\newoverline{v}mu\right)\left(z_0 + \lambda\right)\right)\approx\frac{1}{6}\frac{\partial\left(n\newoverline{v}mu\right)}{\partial z}\left(-2\lambda\right)\nonumber\\ &= -\frac{\lambda, mn\newoverline{v}}{3}\frac{\partial u}{\partial z}, \end{align} \]
where $\newoverline{v}$ and $n$ are assumed to be homogeneous. If the velocity field $u\left(z\right)$ is between two horizontal plates, where the lower one is stationary and the upper one is moved with a constant, positive speed in the x-direction, then, per area $A$, a force
\[ \begin{align} \frac{F}{A} = \eta\frac{\partial u}{\partial z} \end{align} \]
must be applied in order to maintain the flow field. This equation defines the dynamic viscosity $\eta$. The horizontal momentum here flows in the $-z$-direction; at stationarity, one thus has
\[ \begin{align} \eta \approx \frac{n\lambda\newoverline{v}m}{3}.\tag{5.215}\label{eq:dyn_viscosity_kinetic_model} \end{align} \]
Internal friction in a fluid is therefore diffusive momentum transport due to viscosity. If $c$ is the heat capacity per particle, $cT$ is the heat energy per particle. Setting $q = cT$, one obtains
\[ \begin{align} j_z &= \frac{\text{heat}}{\text{time}\times\text{area}} = \frac{1}{6}\left(\left(n\newoverline{v}cT\right)\left(z_0 - \lambda\right) - \left(n\newoverline{v}cT\right)\left(z_0 + \lambda\right)\right)\approx\frac{1}{6}\frac{\partial\left(n\newoverline{v}cT\right)}{\partial z}\left(-2\lambda\right)\nonumber\\ &= -\frac{\lambda cn\newoverline{v}}{3}\frac{\partial T}{\partial z}, \end{align} \]
The quantity
\[ \begin{align} \kappa\approx\frac{\lambda\newoverline{v}}{3} \end{align} \]
is called the thermal diffusivity, which in the kinetic gas model is equal to the diffusion constant. Thus one has
\[ \begin{align} j_z &= -cn\kappa\frac{\partial T}{\partial z} \end{align} \]
This can be generalized three-dimensionally to
\[ \begin{align} \mathbf{j}_q = -\rho c_s\kappa\nabla T\tag{5.219}\label{eq:heat_current_from_heat_conduction} \end{align} \]
where the heat capacity per particle $c$ has been replaced by the specific heat capacity $c_s$. A formal continuity equation also applies to heat conduction, as long as heat conduction constitutes the only heat power density:
\[ \begin{align} \frac{\partial\left(ncT\right)}{\partial t} + \nabla\cdot\mathbf{j}_q &= 0\tag{5.220}\label{eq:heat_conduction_equation_pre} \end{align} \]
This can be generalized to
where $\newtilde{I}$ is the internal energy density. This is the heat conduction equation. If $n$, $c$ and $\kappa$ are independent of time and space, Eq. (5.220) simplifies to
\[ \begin{align} \frac{\partial T}{\partial t} &= \kappa\Delta T. \end{align} \]
5.4.1 Clausius-Clapeyron equation
A phase is a macroscopic manifestation of matter, for example solid, liquid or gaseous. However, even within a solid there can be different phases that differ, for example, in their magnetic properties or lattice structures. First of all, a two-phase equilibrium is assumed, for example consisting of liquid water and water vapor. The liquid phase is denoted by 1, the gaseous phase by 2. The entire system is in thermodynamic equilibrium, so that $\left(p, T\right)$ is the same in both systems. The subsystems are open, so particle exchange takes place. The equilibrium condition according to Eq. (5.54) is then
\[ \begin{align} \mu_1\left(T, p\right) = \mu_2\left(T, p\right),\tag{5.223}\label{eq:cond_chemical_pot_equilibrium} \end{align} \]
where $\mu$ denotes the chemical potential. We are now interested in the pressure $p_S\left(T\right)$ at which both phases coexist. This means
\[ \begin{align} \mu_1\left(T, p_S\left(T\right)\right) = \mu_2\left(T, p_S\left(T\right)\right). \end{align} \]
This is an implicit equation for the unknown function $p_S\left(T\right)$. One differentiates the equation totally with respect to $T$:
\[ \begin{align} \frac{\partial\mu_1\left(T, p\right)}{\partial T} + \frac{\partial\mu_1\left(T, p\right)}{\partial p}\frac{dp_S\left(T\right)}{d T} = \frac{\partial\mu_2\left(T, p\right)}{\partial T} + \frac{\partial\mu_2\left(T, p\right)}{\partial p}\frac{dp_S\left(T\right)}{dT} \end{align} \]
Now one inserts Eqs. (5.105) and (5.107):
\[ \begin{align} - \frac{S_1}{N_1} + \frac{V_1}{N_1}\frac{dp_S}{dT} = -\frac{S_2}{N_2} + \frac{V_2}{N_2}\frac{dp_S}{dT} \end{align} \]
For a state variable $Z$, one has
\[ \begin{align} \frac{Z_i}{N_i} = \frac{Z_i}{m_i}\frac{m_i}{N_i} = z_iM_i \end{align} \]
with mass $m_i$ and specific quantity
\[ \begin{align} z_i \coloneqq \frac{Z_i}{m_i}. \end{align} \]
This results in the definitions $\Delta s \coloneqq s_2 - s_1$ and $\Delta v \coloneqq v_2 - v_1$
\[ \begin{align} \Delta s = \Delta v\frac{dp_S}{dT}. \end{align} \]
One can further use the specific phase-transition heat
\[ \begin{align} c \coloneqq T\Delta s \end{align} \]
:
This is the Clausius-Clapeyron equation. This equation can be solved exactly for simple functions $c = c\left(T\right)$. For the volume per particle $v$, one has
\[ \begin{align} v = \frac{V}{N} = \frac{V}{nN_A} = \frac{VM}{MnN_A} = \frac{M}{N_A\rho}, \end{align} \]
This can be applied to a liquid, for example, with the density $\rho = \rho_l$. For an ideal gas, one has
\[ \begin{align} v = \frac{R_sT}{p} = \frac{R_sT}{p_S}, \end{align} \]
it thus follows
\[ \begin{align} T\Delta v = \frac{R_sT^2}{p_S} - \frac{MT}{N_A\rho_l}\Rightarrow\frac{1}{T\Delta v} = \frac{p_SN_A\rho_l}{N_A\rho_lR_sT^2 - p_SMT}. \end{align} \]
Thus, in the case of a liquid and an ideal gas, one can write
\[ \begin{align} \frac{dp_S}{dT} = \frac{cp_SN_A\rho_l}{N_A\rho_lR_sT^2 - p_SMT}. \end{align} \]
In meteorology, the approximation
\[ \begin{align} T\Delta v\approx\frac{R_sT^2}{p_S} \end{align} \]
is often made; it then follows
\[ \begin{align} \frac{1}{p_S}\frac{dp_S}{dT} = \frac{c}{R_sT^2}.\tag{5.237}\label{eq:clausius-clapeyron_vereinfacht} \end{align} \]
In the case of a temperature-independent phase transition enthalpy $c$, this can be solved analytically, in which case the following holds
\[ \begin{align} p_S = p_S\left(T\right) = k\exp\left(-\frac{T_0}{T}\right), \end{align} \]
for it then follows
\[ \begin{align} \frac{dp_S}{dT} = \frac{T_0}{T^2}p_S. \end{align} \]
This implies
\[ \begin{align} T_0 = \frac{c}{R_s}, \end{align} \]
the constant $k$ remains undetermined at this point.
With regard to the atmosphere, the saturation vapor pressure curve of water is of particular interest. Since the heat of vaporization or sublimation of water is temperature-dependent in a way that cannot be precisely determined analytically, there is no exact expression for the saturation vapor pressure curve for water. The formulas recommended in [41] can help here. Over liquid water, for $T\in\left[-45, 60\right]^\circ$ C, one has
\[ \begin{align} e\left(t\right) = 6, 112\exp\left(\frac{17, 62t}{243, 12 + t}\right), \end{align} \]
over ice, for $T\in\left[-65, 0\right]^\circ$ C, one has
\[ \begin{align} e\left(t\right) = 6,112\exp\left(\frac{22, 46t}{272, 62 + t}\right), \end{align} \]
where $t$ denotes the temperature in $^\circ$C and $e$ denotes the respective saturation vapor pressure in hPa.
5.4.2 Osmotic pressure
We assume a solution of a substance $B$ in a substance $A$, where the substances do not interact with each other. For the partition function $\Omega$ of the system, one then has
\[ \begin{align} \Omega = \Omega_A\Omega_B. \end{align} \]
For the entropy $S$ of the system, it follows
\[ \begin{align} S = k_B\ln\left(\Omega\right) = k_B\ln\left(\Omega_A\Omega_B\right) = k_B\ln\left(\Omega_A\right) + k_B\ln\left(\Omega_B\right). \end{align} \]
From Eq. (5.142), it follows
\[ \begin{align} \Omega_B \propto V^{N_B} \Rightarrow S = S_A + k_BN_B\ln\left(V\right) + f\left(E, N_B\right) \end{align} \]
with a function $f = f\left(E, N_B\right)$, which does not depend on the volume. For the pressure $p$ of the system, it follows
\[ \begin{align} p \stackrel{\href{#eq:pressure_prop_0}{\text{Eq. (5.46)}}}{=} T\left(\frac{\partial S}{\partial V}\right)_E = T\left(\frac{\partial S_A}{\partial V}\right)_E + T\frac{k_BN_B}{V} = p_A + T\frac{k_BN_B}{V} =: p_A + p_\text{osm}. \end{align} \]
The pressure difference $p_\text{osm}$ compared to the pure phase $A$ is called osmotic pressure, for which the following holds
with the concentration $n_B\coloneqq\frac{N_B}{V}$ of the solute. Eq. (5.247) is called van't Hoff's law.
5.4.3 Hydraulic potential
The hydraulic potential is used to understand the mass flux in media driven by pressure gradients and gravity. From Eq. (5.105) it follows that
\[ \begin{align} \mu &= \mu_0 + \int_{p_0}^p\left(\frac{\partial\mu}{\partial p'}\right)_{N, S}dp' = \mu_0 + \int_{p_0}^p\frac{V}{N}dp' = \mu_0 + \frac{V}{N}\int_{p_0}^pdp' = \mu_0 + \frac{\left(p - p_0\right)V}{N} = \mu_0 + \frac{\left(p - p_0\right)M}{\rho}\nonumber\\ \Leftrightarrow\mu &= \mu_0 - \frac{p_0M}{\rho} + \frac{pM}{\rho}\nonumber\\ \Leftrightarrow\frac{\mu}{M} &= \frac{\mu_0}{M} - \frac{p_0}{\rho} + \frac{p}{\rho}. \end{align} \]
One now performs the relabelings
\[ \begin{align} \frac{\mu}{M} &\to \mu,\nonumber\\ \frac{\mu_0}{M} - \frac{p_0}{\rho} &\to \mu_0\nonumber \end{align} \]
so that it follows that
\[ \begin{align} \mu &= \mu_0 + \frac{p}{\rho}. \end{align} \]
Gravity is not yet included here. The potential energy reads $E_\text{pot} = NMgz$, hence
\[ \begin{align} \left(\frac{\partial E_\text{pot}}{M\partial N}\right)_{S, V} &= gz. \end{align} \]
With Eq. (5.91) one now obtains
which is the equation for the hydraulic potential. The quantity $z$ is also referred to as the piezometric head.
5.4.3.1 Darcy's law
Taking the spatial gradient of Eq. (5.251), one obtains
\[ \begin{align} -\nabla\mu &= -\nabla\left(\frac{p}{\rho} + gz\right). \end{align} \]
This has the dimension of a mass flux density $\mathbf{j}$:
This is Darcy's law. The material-dependent quantity $\kappa$ is called the hydraulic conductivity; its SI unit is $\frac{\text{kg}\cdot\text{s}}{\text{m}^3}$.
5.4.4 Vapor pressure over a solution
At a given pressure $p$, the phase transition occurs at a temperature $T_s$ between phase $A$, which is liquid (e.g. water), and phase $C$, which is gaseous or solid. This is described by a function $p_S = p_S\left(T_s\right)$, which is a solution of the Clausius-Clapeyron equation, Eq. (5.231). If one now dissolves a substance $B$ in $A$ (e.g. salt), which is confined to $A$, $T_s$ shifts by $\Delta T_s$. Denote
\[ \begin{align} c\coloneqq\frac{N_B}{N_A} = \frac{N_B/V}{N_A/V} = \frac{n_B}{n_A} \end{align} \]
as the ratio of the concentrations of solute and solvent. For the chemical potential $\mu_A = \mu_A\left(T, p_A\right)$, one has
\[ \begin{align} \mu_A\left(T, p_A\right) &= \mu_A\left(T, p - p_\text{osm}\right) \stackrel{p_\text{osm} \ll p}{\approx} \mu_A\left(T, p\right) - p_\text{osm}\left(\frac{\partial\mu}{\partial p}\right)_T \stackrel{\href{#eq:vant_hoff_law}{\text{Eq. (5.247)}}}{=} \mu_A\left(T, p\right) - k_BTn_B\left(\frac{\partial\mu}{\partial p}\right)_T\nonumber\\ & \stackrel{\href{#eq:maxwell_rel_3}{\text{Eq. (5.108)}}}{=} \mu_A\left(T, p\right) - k_BTn_B\frac{V}{N_A} = \mu_A\left(T, p\right) - ck_BT\tag{5.255}\label{eq:salt_sat_deriv_1}. \end{align} \]
Within the solution, one has
\[ \begin{align} \mu_{A, B}\left(T, p\right) = \mu_A\left(T, p_A\right) \stackrel{\href{#eq:salt_sat_deriv_1}{\text{Eq. (5.255)}}}{\approx} \mu_A\left(T, p\right) - ck_BT. \end{align} \]
Now an equilibrium condition is needed. Since one is dealing with particle exchange, the chemical potential of the solution $A$, $B$ must be equated with that of the other phase $C$:
\[ \begin{align} \mu_{A, B}\left(T, p\right) &= \mu_C\left(T, p\right)\nonumber\\ \stackrel{T = T_s + \Delta T_s}{\Leftrightarrow} \mu_{A, B}\left(T_s + \Delta T_s, p\right) &= \mu_C\left(T_s + \Delta T_s, p\right)\nonumber\\ \stackrel{\href{#eq:salt_sat_deriv_1}{\text{Eq. (5.255)}}}{\Leftrightarrow} \mu_A\left(T_s + \Delta T_s, p\right) - ck_BT &= \mu_C\left(T_s + \Delta T_s, p\right)\tag{5.257}\label{eq:salt_sat_deriv_0} \end{align} \]
From Eq. (5.128), it follows that
\[ \begin{align} \left(\frac{\partial\mu_A}{\partial T}\right)_p = -s_A, \end{align} \]
which implies
\[ \begin{align} \mu_A\left(T_s + \Delta T_s, p\right) \approx \mu_A\left(T_s, p\right) - s_A\Delta T_s \end{align} \]
The same applies to phase $C$
\[ \begin{align} \mu_C\left(T_s + \Delta T_s, p\right) = \mu_C\left(T_s, p\right) - s_C\Delta T_s. \end{align} \]
Substituting the last two equations into Eq. (5.257), one obtains
\[ \begin{align} \mu_A\left(T_s, p\right) - s_A\Delta T_s - ck_BT_s = \mu_C\left(T_s, p\right) - s_C\Delta T_s. \end{align} \]
In the last step, a small term $ck_B\Delta T_s$ was omitted. At $c = 0$, one has
\[ \begin{align} \mu_A\left(T_s, p\right) = \mu_C\left(T_s, p\right),\tag{5.262}\label{eq:salt_sat_deriv_3} \end{align} \]
which corresponds to the phase boundary in the absence of the solute. Thus, it follows
\[ \begin{align} -s_A\Delta T_s - ck_BT_s &= -s_C\Delta T_s\nonumber\\ \Leftrightarrow \Delta T_s&= c\frac{k_BT_s}{s_C - s_A}. \end{align} \]
For the entropy difference, one has
\[ \begin{align} s_C - s_A = \frac{L_{A\to C}}{T_s}, \end{align} \]
here $L_{A\to C}$ is the phase transition enthalpy in the transition from $A$ to $C$. Thus one obtains
\[ \begin{align} \Delta T_s &= c\frac{k_BT_s^2}{L_{A\to C}}. \end{align} \]
Two cases can be distinguished here:
$C$ is gaseous: This implies $L_{A\to C} > 0$, which in turn leads to $\Delta T_s > 0$, which corresponds to a boiling point increase. This is the case, for example, over the ocean surface.
$C$ is solid: This implies $L_{A\to C} < 0$, which in turn leads to $\Delta T_s < 0$, which corresponds to a freezing point depression. This reduces the freezing point of ocean water to below zero degrees Celsius.
Instead of the change in boiling or freezing point, the influence of a solution on the saturation vapor pressure can also be determined. Instead of Eq. (5.257), one starts here with
\[ \begin{align} \mu_A\left(T, p_s + \Delta p_s\right) - ck_BT &= \mu_C\left(T, p_s + \Delta p_s\right)\tag{5.266}\label{eq:salt_sat_deriv_2} \end{align} \]
From Eq. (5.128), it follows that
\[ \begin{align} \left(\frac{\partial\mu_A}{\partial p}\right)_T = v_A, \end{align} \]
which implies
\[ \begin{align} \mu_A\left(T, p_s + \Delta p_s\right) = \mu_A\left(T, p_s\right) + \int_{p_s}^{p_s + \Delta p_s}\left(\frac{\partial\mu_A}{\partial p}\right)_Tdp = \mu_A\left(T, p_s\right) + \int_{p_s}^{p_s + \Delta p_s}v_Adp \end{align} \]
The same applies to phase $C$
\[ \begin{align} \mu_C\left(T, p_s + \Delta p_s\right) = \mu_C\left(T, p_s\right) + \int_{p_s}^{p_s + \Delta p_s}\left(\frac{\partial\mu_C}{\partial p}\right)_Tdp = \mu_C\left(T, p_s\right) + \int_{p_s}^{p_s + \Delta p_s}v_Cdp. \end{align} \]
Substituting the last two equations into Eq. (5.266), one obtains
\[ \begin{align} \mu_A\left(T, p_s\right) + \int_{p_s}^{p_s + \Delta p_s}v_Adp - ck_BT = \mu_C\left(T, p_s\right) + \int_{p_s}^{p_s + \Delta p_s}v_Cdp. \end{align} \]
Analogously to Eq. (5.262), one has
\[ \begin{align} \mu_A\left(T, p_s\right) = \mu_C\left(T, p_s\right), \end{align} \]
it follows from this
\[ \begin{align} \int_{p_s}^{p_s + \Delta p_s}v_Adp - ck_BT = \int_{p_s}^{p_s + \Delta p_s}v_Cdp \Rightarrow \int_{p_s}^{p_s + \Delta p_s}\frac{dp}{n_A} - ck_BT = \int_{p_s}^{p_s + \Delta p_s}\frac{dp_s}{n_C}. \end{align} \]
In this case, since $A$ is liquid and $C$ is gaseous, $n_A\gg n_C$ holds, which leads to
\[ \begin{align} -ck_BT \approx \int_{p_s}^{p_s + \Delta p_s}\frac{dp}{n_C} \end{align} \]
With the equation of state of ideal gases, it follows
\[ \begin{align} -ck_BT &= k_BT\int_{p_s}^{p_s + \Delta p_s}\frac{dp}{p_s} = k_BT\ln\left(\frac{p_s + \Delta p_s}{p_s}\right)\nonumber \end{align} \]
In the case of small concentrations $c\ll 1$ one obtains approximately
\[ \begin{align} \frac{\Delta p_s}{p_s} = -c, \end{align} \]
which is called Raoult's law.
5.4.5 Boltzmann equation
This section aims to establish a connection between statistical physics and hydrodynamics. The central quantity here is the function
\[ \begin{align} f = f\left(\mathbf{r}, \mathbf{v}, t\right), \end{align} \]
this is the probability density for encountering particles at time $t$ in the phase-space volume at $\mathbf{r}$, $\mathbf{v}$; here one has
\[ \begin{align} N = \int_{\mathbb{R}^6}f\left(\mathbf{r}, \mathbf{v}, t\right)d^3vd^3r \end{align} \]
with $N$ as the particle number. The total derivative of $f$ reads $\left(\frac{\partial}{\partial t} + \mathbf{v}\cdot\nabla_\mathbf{r} + \frac{\mathbf{F}}{m}\cdot\nabla_\mathbf{v}\right)f\left(\mathbf{r}, \mathbf{v}, t\right)$; the equation
is called the collisionless Boltzmann equation. Here $\md{\mathbf{v}} = \frac{\mathbf{F}}{m}$ with $\mathbf{F}$ as the sum of external forces (electromagnetic field, gravity, apparent forces) was used. Internal interactions caused by particle collisions (pressure gradient acceleration, viscosity) have not yet been taken into account; a term $F_\text{int}$ is used for them. This term will now be developed step by step. It should describe how
particles are scattered into a small phase space volume at $\left(\mathbf{r}, \mathbf{v}\right)$,
particles are scattered out of a small phase space volume at $\left(\mathbf{r}, \mathbf{v}\right)$.
First, it is expected that $F_\text{int}$ is an integral over all velocities:
\[ \begin{align} F_\text{int} = \int_{\mathbb{R}^3}\dots d^3v_1 \end{align} \]
A position integral is not useful because collisions only take place locally. Furthermore, it is expected that the loss of particles in the phase space volume $\left(\mathbf{r}, \mathbf{v}\right)$ is proportional to $f\left(\mathbf{r}, \mathbf{v}, t\right)$, i.e.
\[ \begin{align} F_\text{int} = \int_{\mathbb{R}^3}-f\left(\mathbf{r}, \mathbf{v}, t\right) + \dots d^3v_1 \end{align} \]
At the point $\mathbf{v}_1$, collisions with particles in a small phase space volume at $\left(\mathbf{r}, \mathbf{v}_1\right)$ are considered, so one continues
\[ \begin{align} F_\text{int} = \int_{\mathbb{R}^3}-f\left(\mathbf{r}, \mathbf{v}, t\right)f\left(\mathbf{r}, \mathbf{v}_1, t\right) + \dots d^3v_1. \end{align} \]
The number of collisions per time is proportional to the relative velocity $\mathbf{V}\coloneqq\mathbf{v} - \mathbf{v}_1$, one obtains
\[ \begin{align} F_\text{int} = \int_{\mathbb{R}^3}-f\left(\mathbf{r}, \mathbf{v}, t\right)f\left(\mathbf{r}, \mathbf{v}_1, t\right)\left|\mathbf{V}\right| + \dots d^3v_1. \end{align} \]
The current density itself is not enough to determine the number of scattering processes per time: if all particles are ideal mass points, for example, there will be no collisions at all. This is described by the cross section $\sigma$, which is why one makes the ansatz
\[ \begin{align} F_\text{int} = \int_{\mathbb{R}^3}-f\left(\mathbf{r}, \mathbf{v}, t\right)f\left(\mathbf{r}, \mathbf{v}_1, t\right)\left|\mathbf{V}\right|\sigma + \dots d^3v_1. \end{align} \]
The velocities of the particles before the collision are $\mathbf{v}$ and $\mathbf{v}_1$, those after the collision are denoted by $\mathbf{v}'$ and $\mathbf{v}_1'$. One further defines the relative velocity after the collision by $\mathbf{V}' \coloneqq \mathbf{v}_1' - \mathbf{v}'$, where $\left|\mathbf{V}\right| = \left|\mathbf{V}'\right|$ holds. Denote by $\Omega$ the solid angle between $\mathbf{V}$ and $\mathbf{V}'$. Particles are scattered into the solid angle element $d\Omega$ in proportion to $\frac{d\sigma}{d\Omega}d\Omega$, this leads to
\[ \begin{align} F_\text{int} = \int_{\mathbb{R}^3}\int_\Omega-f\left(\mathbf{r}, \mathbf{v}, t\right)f\left(\mathbf{r}, \mathbf{v}_1, t\right)\left|\mathbf{V}\right|\frac{d\sigma}{d\Omega}d\Omega + \dots d^3v_1. \end{align} \]
Analogously, a scattering process from $\mathbf{v}'$ and $\mathbf{v}_1'$ to $\mathbf{v}$ and $\mathbf{v}_1$ takes place; this leads to
\[ \begin{align} F_\text{int} = \int_{\mathbb{R}^3}\int_\Omega\left[f\left(\mathbf{r}, \mathbf{v}', t\right)f\left(\mathbf{r}, \mathbf{v}_1', t\right) - f\left(\mathbf{r}, \mathbf{v}, t\right)f\left(\mathbf{r}, \mathbf{v}_1, t\right)\right]\left|\mathbf{V}\right|\frac{d\sigma}{d\Omega}d\Omega d^3v_1. \end{align} \]
The quantities $\mathbf{v}'$ and $\mathbf{v}_1'$ are determined by the scattering problem ($\mathbf{v}$, $\mathbf{v}_1$, $\Omega$ as well as conservation of momentum and energy). Writing this on the right-hand side of Eq. (5.278), one obtains
\[ \begin{align} &\left(\frac{\partial}{\partial t} + \mathbf{v}\cdot\nabla_\mathbf{r} + \frac{\mathbf{F}}{m}\cdot\nabla_\mathbf{v}\right)f\left(\mathbf{r}, \mathbf{v}, t\right)\nonumber\\ &= \int_{\mathbb{R}^3}\int_\Omega\left[f\left(\mathbf{r}, \mathbf{v}', t\right)f\left(\mathbf{r}, \mathbf{v}_1', t\right) - f\left(\mathbf{r}, \mathbf{v}, t\right)f\left(\mathbf{r}, \mathbf{v}_1, t\right)\right]\left|\mathbf{V}\right|\frac{d\sigma}{d\Omega}d\Omega d^3v_1. \end{align} \]
This is the Boltzmann equation. It can be regarded as the classical analogue of the master equation, Eq. (5.17). It is important to note that no assumptions were made about the nature of the interactions that lead to the collisions. This is implicit in the cross section $\frac{d\sigma}{d\Omega}$.
The central quantities of hydrodynamics can be written in terms of $f$:
\[ \begin{align} N &= \int_{\mathbb{R}^6}f\left(\mathbf{r}, \mathbf{v}', t\right)d^3v'd^3r \hastobe \int_{\mathbb{R}^3}n\left(\mathbf{r}, t\right)d^3v' \Rightarrow n\left(\mathbf{r}, t\right) = \int_{\mathbb{R}^3}f\left(\mathbf{r}, \mathbf{v}', t\right)d^3v',\nonumber\\ \rho\left(\mathbf{r}, t\right) &= mn\left(\mathbf{r}, t\right) = \int_{\mathbb{R}^3}mf\left(\mathbf{r}, \mathbf{v}', t\right)d^3v',\nonumber\\ \rho\left(\mathbf{r}, t\right)\mathbf{v}\left(\mathbf{r}, t\right) &= \int_{\mathbb{R}^3}mf\left(\mathbf{r}, \mathbf{v}', t\right)\mathbf{v}'d^3v',\nonumber\\ \mathbf{v}\left(\mathbf{r}, t\right) &= \frac{\rho\left(\mathbf{r}, t\right)\mathbf{v}\left(\mathbf{r}, t\right)}{\rho\left(\mathbf{r}, t\right)} = \frac{\int_{\mathbb{R}^3}mf\left(\mathbf{r}, \mathbf{v}', t\right)\mathbf{v}'d^3v'}{\int_{\mathbb{R}^3}mf\left(\mathbf{r}, \mathbf{v}', t\right)d^3v'} = \frac{\int_{\mathbb{R}^3}f\left(\mathbf{r}, \mathbf{v}', t\right)\mathbf{v}'d^3v'}{\int_{\mathbb{R}^3}f\left(\mathbf{r}, \mathbf{v}', t\right)d^3v'} \end{align} \]
5.5 Capillarity
Consider an interface $A$ coinciding with $\mathbb{R}^2$. Expanding it by a factor $d\epsilon$ in the $x$-direction can be interpreted as applying the map
\[ \begin{align} \left(x, y\right)^T \mapsto \left(x\left(1 + d\epsilon\right), y\right)^T \end{align} \]
to the interface. In doing so, the energy of the system increases by
\[ \begin{align} dU &= A\sigma d\epsilon \end{align} \]
where the proportionality constant $\sigma$ is the mechanical surface tension. Hence
\[ \begin{align} \sigma = \frac{1}{A}\frac{dU}{d\epsilon}. \end{align} \]
Now consider a cylindrical disk of radius $r$ and opening angle $\varphi$, lying in $\mathbb{R}^2$:
\[ \begin{align} Z \coloneqq \left\{\left(x, y, z\right)^T\in\mathbb{R}^3\newvline\left|x\right|\leq r\sin\left(\varphi\right)\land 0\leq z\leq\sqrt{r^2-x^2} - r\left(1-\cos\left(\varphi\right)\right)\right\} \end{align} \]
The force acting on a surface element $dA = r2\sin\left(\varphi\right)dy$ in the $z$-direction is
\[ \begin{align} F_z = -2dy\sigma\sin\left(\varphi\right). \end{align} \]
In this case, the sign of $r$ is negative, and the capillary pressure $p_r$ generated by $F_z$ is therefore positive. Thus
\[ \begin{align} p_r = -\frac{\sigma}{r}. \end{align} \]
For a surface displacement $\eta$, Eq. (B.43) implies
\[ \begin{align} p_k \approx -\sigma\frac{\partial^2\eta}{\partial x^2}. \end{align} \]
So far, only the curvature in one spatial direction has been considered. If both are taken into account, one obtains
5.5.1 Kelvin equation
Up to this point, it has been assumed that the phase boundary is flat or that the surface tension is zero. In general, the equilibrium condition Eq. (5.223) depends on the radius $R$ of the interface:
\[ \begin{align} \mu_1\left(R, T, V\right) = \mu_2\left(R, T, V\right)\tag{5.296}\label{eq:cond_chemical_pot_equilibrium_r} \end{align} \]
Here the dependence on $p$ has been replaced by a dependence on $V$. In what follows, only $R$ is carried as an argument, since $T$ and $V$ are treated as constants. For the particle number density $n = N/V$, Eq. (5.111) gives
\[ \begin{align} n = \left(\frac{\partial p}{\partial\mu}\right)_{T, V} \stackrel{T\text{, }V\text{ const.}}{\Rightarrow} d\mu = \frac{dp}{n}.\tag{5.297}\label{eq:kelvin_eq_deriv_0} \end{align} \]
Eq. (5.296) implies
\[ \begin{align} \mu_1\left(R\right) - \mu_1\left(\infty\right) &= \mu_2\left(R\right) - \mu_2\left(\infty\right)\nonumber\\ \Leftrightarrow\int_\infty^R\frac{d\mu_1\left(r\right)}{dr}dr &= \int_\infty^R\frac{d\mu_2\left(r\right)}{dr}dr. \end{align} \]
This is now rewritten using $p_i = p_i\left(r_i\right)$:
\[ \begin{align} \int_{p_1\left(\infty\right)}^{p_1\left(R\right)}\frac{d\mu_1\left(r_1\left(p_1\right)\right)}{dr_1}\frac{dr_1}{dp_1}dr_1 &= \int_{p_2\left(\infty\right)}^{p_2\left(R\right)}\frac{d\mu_2\left(r_2\left(p_2\right)\right)}{dr_2}\frac{dr_2}{dp_2}dr_2\nonumber\\ \Leftrightarrow\int_{p_1\left(\infty\right)}^{p_1\left(R\right)}\frac{d\mu_1\left(p_1\right)}{dp_1}dp_1 &= \int_{p_2\left(\infty\right)}^{p_2\left(R\right)}\frac{d\mu_2\left(p_2\right)}{dp_2}dp_2 \end{align} \]
One has
\[ \begin{align} p_1\left(\infty\right) = p_2\left(\infty\right) = p_S. \end{align} \]
As before, 1 denotes the liquid phase and 2 the gas phase. Define
\[ \begin{align} p_R \coloneqq p_2\left(R\right). \end{align} \]
From Eq. (5.295), it follows that
\[ \begin{align} p_1\left(R\right) = p_R + \frac{2\gamma}{R}. \end{align} \]
Hence
\[ \begin{align} \int_{p_S}^{p_R + \frac{2\gamma}{R}}\frac{d\mu_1\left(p_1\right)}{dp_1}dp_1 &= \int_{p_S}^{p_R}\frac{d\mu_2\left(p_2\right)}{dp_2}dp_2. \end{align} \]
Substituting Eq. (5.297) yields
\[ \begin{align} \int_{p_S}^{p_R + \frac{2\gamma}{R}}\frac{1}{n_1\left(p_1\right)}dp_1 &= \int_{p_S}^{p_R}\frac{1}{n_2\left(p_2\right)}dp_2. \end{align} \]
If the liquid phase is assumed incompressible, then $n_1 = n_l > 0$ is constant and independent of $p$. With the ideal-gas equation of state $p_2 = n_2k_BT$, one obtains
\[ \begin{align} \frac{p_R + \frac{2\gamma}{R} - p_S}{n_l} = k_BT\int_{p_S}^{p_R}\frac{1}{p_2}dp_2 = k_BT\ln\left(\frac{p_R}{p_S}\right)\nonumber\\ \Leftrightarrow \frac{N_A}{M}k_BT\ln\left(\frac{p_R}{p_S}\right) = \frac{N_A}{M}\frac{p_R + \frac{2\gamma}{R} - p_S}{n_l}\nonumber\\ \Leftrightarrow R_sT\ln\left(\frac{p_R}{p_S}\right) = \frac{N_A}{M}\frac{p_R + \frac{2\gamma}{R} - p_S}{n_l}, \end{align} \]
where $M$ is the molar mass of the substance under consideration. Under the assumption
\[ \begin{align} \frac{2\gamma}{R} \gg p_R - p_S \end{align} \]
this yields the Kelvin equation
The quantity
\[ \begin{align} \Delta U \coloneqq \frac{p_R}{p_S} - 1 = \exp\left(\frac{2\gamma}{R_sTR\rho_l}\right) - 1 \geq 0 \end{align} \]
is the saturation humidity over a curved surface relative to the saturation humidity over a flat surface. Fig. 5.1 illustrates this for H$_2$O.
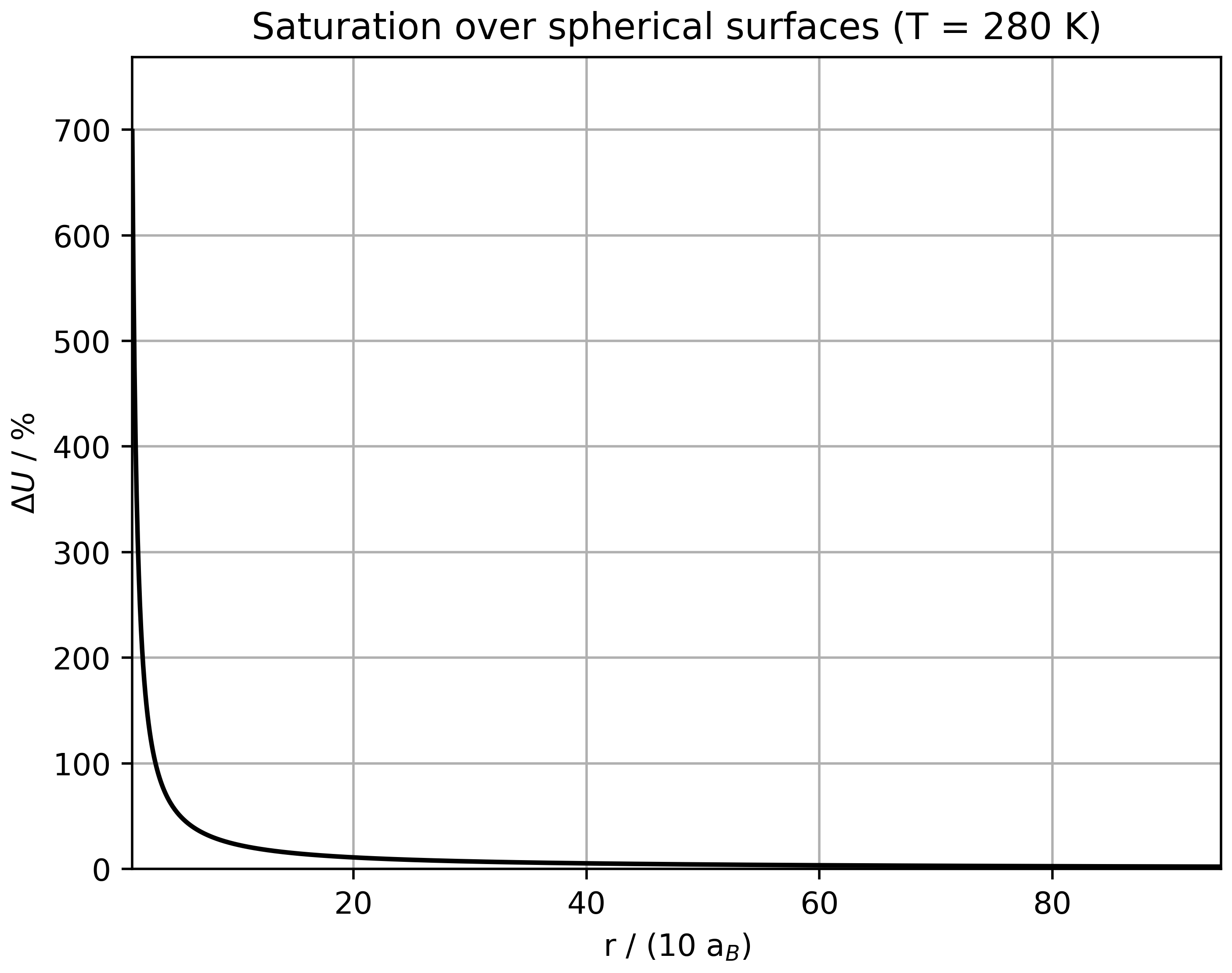
5.6 Photon gas
Consider a cavity of temperature $T$ in equilibrium with electromagnetic radiation. We seek the spectral energy density $u\left(\omega\right)$ in the cavity. The properties of electromagnetic waves were discussed in Sect. 3.3. These are transverse waves. If the wave vector points in the $z$-direction, $\mathbf{k} = k\mathbf{e}_z$, the electric field for standing waves is
\[ \begin{align} \mathbf{E}\left(z, t\right) = \left(E_{0, x}\mathbf{e}_x + E_{0, y}\mathbf{e}_y\right)\sin\left(kz\right)\sin\left(\omega t\right). \end{align} \]
Any phases are neglected. The $\mathbf{B}$ field can be determined from this. Let the cavity be cubic with volume $V = L^3$, and let the walls be metallic. In this case, the tangential component of the electric field vanishes at the surface, since otherwise currents would be induced and energy dissipated. This yields the discrete $k$-values
\[ \begin{align} k_iL = i\pi\Leftrightarrow k_i = \frac{\pi}{L}i \end{align} \]
with $i\in \mathbb{N}$ and $i\geq 1$; negative values of $i$ are excluded because they do not produce new solutions. Sums over $k_i$ values can again be replaced by integrals:
\[ \begin{align} \sum_{k_i}^{}\dotsc = \frac{L}{\pi}\int_0^\infty\dotsc dk = \frac{L}{2\pi}\int_{-\infty}^{\infty}\dotsc dk. \end{align} \]
For a sum over all modes in the cavity, one has
\[ \begin{align} \sum_{m = 1}^{2}\sum_{\mathbf{k}}^{}\dotsc = 2\sum_{\mathbf{k}}^{}\dotsc = 2\left(\frac{L}{2\pi}\right)^3\int\dotsc d^3k = \frac{2V}{\left(2\pi\right)^3}\int\dotsc d^3k. \end{align} \]
$m$ runs over the two possible polarization directions. For the energies $E = E\left(\mathbf{k}, m\right)$, according to Eq. (4.6)
\[ \begin{align} E\left(\mathbf{k}, m\right) = \hbar\omega\left(k\right) = \hbar ck. \end{align} \]
A microstate $r$ of the system is specified by the occupation numbers
\[ \begin{align} r = \left(n_{\mathbf{k}, m}\right) \end{align} \]
that is, the number of excitations for each vibrational mode; one excitation corresponds to one photon. The energy of the microstate is $E_r\left(V\right)$
\[ \begin{align} E_r\left(V\right) = \sum_{m, \mathbf{k}}^{}\hbar\omega\left(k\right)n_{\mathbf{k}, m} = \hbar c\sum_{\mathbf{k}, m}^{}kn_{\mathbf{k}, m}. \end{align} \]
Now the mean occupation numbers $\newoverline{n}_{\mathbf{k}, m}$ are to be determined. To do so, one starts from the grand canonical partition function of a system without a polarization degree of freedom, i.e. $m = 1$:
\[ \begin{align} Y &= \sum_r\exp\left[-\beta\left(E_r - \mu N_r\right)\right] = \sum_{n_{\mathbf{p}_1 = 0}}^{\infty}\exp\left[-\beta\left(E_{p_1} - \mu\right)n_{\mathbf{p}_1}\right]\cdot\sum_{n_{\mathbf{p}_2} = 0}^{\infty}\exp\left[-\beta\left(E_{p_2} - \mu\right)n_{\mathbf{p}_2}\right]\cdot\dotsc\nonumber\\ &\stackrel{\href{ch-39-derivations-of-some-mathematical-formula.html#eq:geometr_reihe}{\text{Eq. (A.8)}}}{=} \prod_{\mathbf{p}}^{}\frac{1}{1 - \exp\left(-\beta\left(E_p - \mu\right)\right)}. \end{align} \]
It follows that
\[ \begin{align} \newoverline{n}_{\mathbf{p}_i} &= \frac{1}{Y}\sum_rn_{\mathbf{p}_i}\exp\left[-\beta\left(E_r - \mu N_r\right)\right] = \frac{1}{Y}\sum_{n_{\mathbf{p}_1} = 0}^{\infty}\dotsc\sum_{n_{\mathbf{p}_i} = 0}^{\infty}\dotsc n_{\mathbf{p}_i}\exp\left[-\beta\left(E_{\mathbf{p}_i} - \mu\right)n_{\mathbf{p}_i}\right]\dotsc\nonumber\\ &= \frac{1}{Y}\sum_{n_{\mathbf{p}_1} = 0}^{\infty}\dotsc\left(\frac{1}{\beta}\frac{\partial}{\partial\mu}\sum_{n_{\mathbf{p}_i} = 0}^{\infty}\exp\left[-\beta\left(E_{\mathbf{p}_i} - \mu\right)n_{\mathbf{p}_i}\right]\right)\dotsc = \frac{1}{Y}\sum_{n_{\mathbf{p}_1} = 0}^{\infty}\dotsc\left(\frac{1}{\beta}\frac{\partial}{\partial\mu}\frac{1}{1 - \exp\left[-\beta\left(E_{\mathbf{p}_i} - \mu\right)\right]}\right)\dotsc\nonumber\\ &= \frac{1}{Y}\sum_{n_{\mathbf{p}_1} = 0}^{\infty}\dotsc\left(\frac{\exp\left[-\beta\left(E_{\mathbf{p}_i} - \mu\right)\right]}{\left(1 - \exp\left[-\beta\left(E_{\mathbf{p}_i} - \mu\right)\right]\right)^2}\right)\dotsc = \frac{\exp\left[-\beta\left(E_{\mathbf{p}_i} - \mu\right)\right]}{1 - \exp\left[-\beta\left(E_{\mathbf{p}_i} - \mu\right)\right]}. \end{align} \]
This is the Bose distribution; it applies to particles with integer spin:
\[ \begin{align} \newoverline{n}_\mathbf{p} = \frac{1}{e^{\beta\left(E_\mathbf{p} - \mu\right)} - 1} \end{align} \]
The energy eigenvalues $E_r\left(V\right)$ do not depend on the number of photons $N$, so
\[ \begin{align} \mu = \newoverline{\frac{\partial E_r\left(V\right)}{\partial N}} = 0, \end{align} \]
the chemical potential vanishes. Therefore
\[ \begin{align} \newoverline{n}_{\mathbf{k}, m} = \frac{1}{\exp\left(\beta E_k\right) - 1}. \end{align} \]
It follows that
\[ \begin{align} E\left(T, V\right) = \newoverline{E_r\left(T, V\right)} &= \sum_{m, \mathbf{k}}^{}E_k\newoverline{n_k} = \frac{2V}{\left(2\pi\right)^3}\int\frac{\hbar ck}{\exp\left(\beta\hbar ck\right) - 1}d^3k = \frac{2V}{\left(2\pi\right)^3}4\pi\int_{0}^{\infty}\frac{\hbar ck^3}{\exp\left(\beta\hbar ck\right) - 1}dk\nonumber\\ &= V\int_{0}^{\infty}\underbrace{\frac{\hbar}{c^3\pi^2}\frac{\omega^3}{\exp\left(\beta\hbar\omega\right) - 1}}_{ = u\left(\omega\right)}d\omega. \end{align} \]
This is the Planck's radiation law. The assumptions of this section are quite specific, but Planck's distribution always applies when matter at temperature $T$ is in equilibrium with electromagnetic radiation. Because of the high propagation speed of electromagnetic waves (the speed of light), this equilibrium is generally justified. Deviations in the spectrum can still occur, for example as absorption lines. Within a cavity in thermal equilibrium, the radiation is isotropic, so the spectral radiance is
\[ \begin{align} J\left(\omega\right) = \frac{u\left(\omega\right)}{4\pi}c = \frac{\hbar}{4\pi^3c^2}\frac{\omega^3}{\exp\left(\frac{\hbar\omega}{k_BT}\right) - 1}. \end{align} \]
The spectral intensity $I\left(\omega\right)$ of a radiating surface is then
\[ \begin{align} I\left(\omega\right) &= \int_{\vartheta = 0}^{\pi/2}\int_{\phi = 0}^{2\pi}J\left(\omega\right)\cos\left(\vartheta\right)\sin\left(\vartheta\right)d\vartheta d\phi = 2\pi\frac{1}{2}J\left(\omega\right) = \frac{\hbar}{4\pi^2c^2}\frac{\omega^3}{\exp\left(\frac{\hbar\omega}{k_BT}\right) - 1}. \end{align} \]
Integrating this over the entire spectrum yields the radiated power density
\[ \begin{align} \frac{P}{A} = \int_{0}^{\infty}I\left(\omega\right)d\omega = \frac{\hbar}{4\pi^2c^2}\int_{0}^{\infty}\frac{\omega^3}{\exp\left(\frac{\hbar\omega}{k_BT}\right) - 1}d\omega = \frac{k_B^4T^4}{4\pi^2c^2\hbar^3}\int_{0}^{\infty}\frac{x^3}{e^x - 1}d^x \end{align} \]
Using Eq. (A.101), one obtains
\[ \begin{align} P = P\left(T\right) = \frac{k_B^4\pi^2}{60c^2\hbar^3}T^4.\tag{5.326}\label{eq:stefan-boltzmann} \end{align} \]
This is the Stefan-Boltzmann law. One defines
\[ \begin{align} \sigma \coloneqq\frac{k_B^4\pi^2}{60c^2\hbar^3} \end{align} \]
as the Stefan-Boltzmann constant. Real bodies at temperature $T$ emit, in the direction specified by $\vartheta$ and $\varphi$ and at angular frequency $\omega$, a spectral radiance that differs from $J\left(\omega, T\right)$ by the factor
\[ \begin{align} \epsilon = \epsilon\left(\omega, T, \vartheta, \varphi\right) \end{align} \]
:
\[ \begin{align} J_{\text{real}}\left(\omega, T, \vartheta, \varphi\right) = \epsilon\left(\omega, T, \vartheta, \varphi\right)J\left(\omega, T\right). \end{align} \]
The factor $\epsilon$ is called emissivity. Consider a small area element $dA$ on the inner surface of the cavity. This element absorbs a power density
\[ \begin{align} \frac{dP_{\text{abs}}}{dA} = \int_{\omega = 0}^\infty\int_{\vartheta = 0}^{\pi/2}\int_{\varphi = 0}^{2\pi}\alpha\left(\omega, T, \vartheta, \varphi\right)J\left(\omega, T\right)\cos\left(\vartheta\right)\sin\left(\vartheta\right)d\varphi d\vartheta d\omega, \end{align} \]
Here the absorption coefficient $\alpha\left(\omega, T, \vartheta, \varphi\right)$ has been introduced. Since thermodynamic equilibrium is assumed, the emitted power density is
\[ \begin{align} \frac{dP_{\text{emt}}}{dA} = \int_{\omega = 0}^\infty\int_{\vartheta = 0}^{\pi/2}\int_{\varphi = 0}^{2\pi}\epsilon\left(\omega, T, \vartheta, \varphi\right)J\left(\omega, T\right)\cos\left(\vartheta\right)\sin\left(\vartheta\right)d\varphi d\vartheta d\omega = \frac{dP_{\text{abs}}}{dA}. \end{align} \]
It follows that
\[ \begin{align} \int_{\omega = 0}^\infty\int_{\vartheta = 0}^{\pi/2}\int_{\varphi = 0}^{2\pi}\left(\epsilon\left(\omega, T, \vartheta, \varphi\right) - \alpha\left(\omega, T, \vartheta, \varphi\right)\right)J\left(\omega, T\right)\cos\left(\vartheta\right)\sin\left(\vartheta\right)d\varphi d\vartheta d\omega = 0. \end{align} \]
Since the spectral energy density of the cavity is constant, one even has
\[ \begin{align} \int_{\vartheta = 0}^{\pi/2}\int_{\varphi = 0}^{2\pi}\left[\epsilon\left(\omega, T, \vartheta, \varphi\right) - \alpha\left(\omega, T, \vartheta, \varphi\right)\right]\sin\left(\vartheta\right)\cos\left(\vartheta\right)d\varphi d\vartheta = 0. \end{align} \]
Define
\[ \begin{align} \epsilon\left(\omega, T\right) \coloneqq\int_{\vartheta = 0}^{\pi/2}\int_{\varphi = 0}^{2\pi}\epsilon\left(\omega, T, \vartheta, \varphi\right)\sin\left(\vartheta\right)\cos\left(\vartheta\right)d\varphi d\vartheta. \end{align} \]
and analogously for the absorption coefficient. Then
\[ \begin{align} \epsilon\left(\omega, T\right) = \alpha\left(\omega, T\right). \end{align} \]
Now consider specifically the direction given by $\vartheta$ and $\varphi$. The spectral radiance emitted by $dA$ is
\[ \begin{align} & J_\text{out}\left(\omega, T\right) = J\left(\omega, T\right)\nonumber\\ &= J\left(\omega, T\right)\left(\epsilon\left(\omega, T, \vartheta, \varphi\right) + \int_{\varphi' = 0}^{2\pi}\int_{\vartheta' = 0}^{\pi/2}s\left(\vartheta', \varphi', \vartheta, \varphi, \omega, T\right)\cos\left(\vartheta'\right)\sin\left(\vartheta'\right)d\vartheta'd\varphi'\right) \end{align} \]
where the expression in parentheses equals one. The scattering cross section $s\left(\vartheta', \varphi', \vartheta, \varphi, \omega, T\right)$ has been introduced. It describes to what extent radiation incident from direction $\left(\vartheta', \varphi'\right)$ is redirected into direction $\left(\vartheta, \varphi\right)$ without intermediate absorption and re-emission. For the incident spectral radiance, one has
\[ \begin{align} & J_\text{in}\left(\omega, T\right) = J\left(\omega, T\right)\nonumber\\ &= J\left(\omega, T\right)\left(\alpha\left(\omega, T, \vartheta, \varphi\right) + \int_{\varphi' = 0}^{2\pi}\int_{\vartheta' = 0}^{\pi/2}s\left(\vartheta, \varphi, \vartheta', \varphi', \omega, T\right)\cos\left(\vartheta'\right)\sin\left(\vartheta'\right)d\vartheta'd\varphi'\right) \end{align} \]
The expression in parentheses must again equal one. Since the Maxwell equations, like the Schrödinger equation, are invariant under time reversal, it follows that
\[ \begin{align} s\left(\vartheta, \varphi, \vartheta', \varphi', \omega, T\right) = s\left(\vartheta', \varphi', \vartheta, \varphi, \omega, T\right). \end{align} \]
Hence
\[ \begin{align} \epsilon\left(\omega, T, \vartheta, \varphi\right) = \alpha\left(\omega, T, \vartheta, \varphi\right) \end{align} \]
This is called Kirchhoff's radiation law. Since a body can never absorb more radiation than is incident upon it, one has $\alpha\leq1$, and therefore
\[ \begin{align} \epsilon\leq1. \end{align} \]
Bodies with $\epsilon\left(\vartheta, \varphi, \omega\right) = 1$ are called black bodies, and the Stefan-Boltzmann law therefore applies only to black bodies.